

Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome
- PMID: 30179222
- PMCID: PMC6159964
- DOI: 10.1172/JCI98688
Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome
Abstract
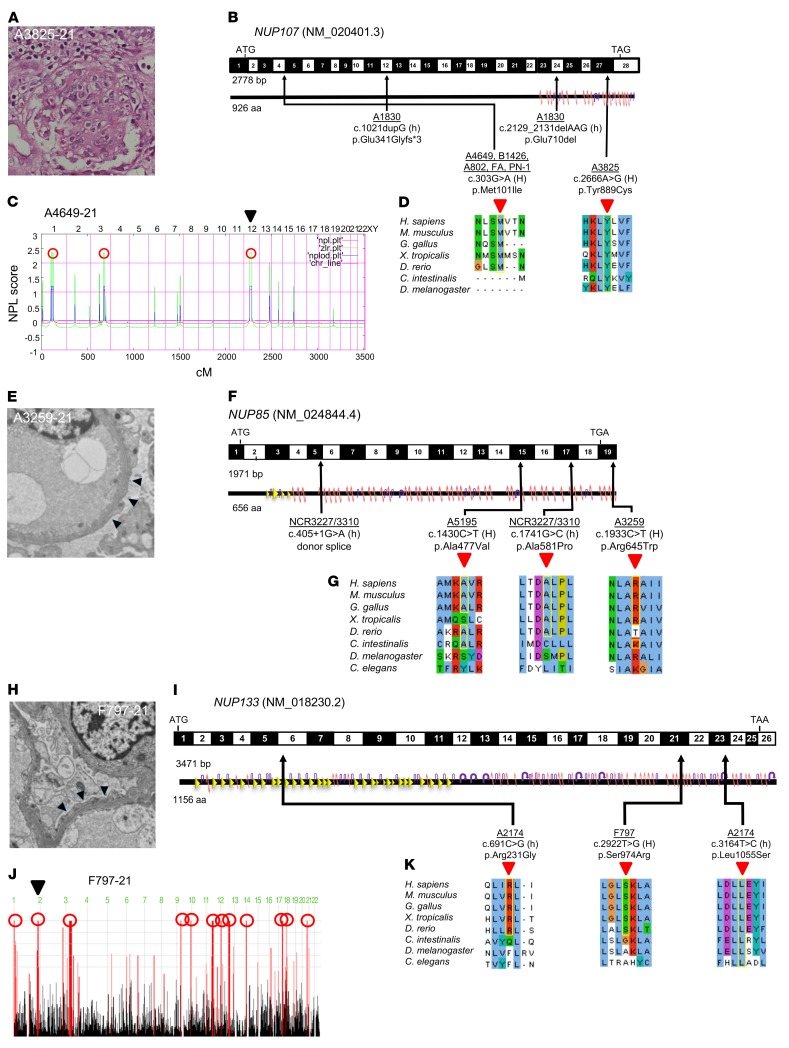
Steroid-resistant nephrotic syndrome (SRNS) almost invariably progresses to end-stage renal disease. Although more than 50 monogenic causes of SRNS have been described, a large proportion of SRNS remains unexplained. Recently, it was discovered that mutations of NUP93 and NUP205, encoding 2 proteins of the inner ring subunit of the nuclear pore complex (NPC), cause SRNS. Here, we describe mutations in genes encoding 4 components of the outer rings of the NPC, namely NUP107, NUP85, NUP133, and NUP160, in 13 families with SRNS. Using coimmunoprecipitation experiments, we showed that certain pathogenic alleles weakened the interaction between neighboring NPC subunits. We demonstrated that morpholino knockdown of nup107, nup85, or nup133 in Xenopus disrupted glomerulogenesis. Re-expression of WT mRNA, but not of mRNA reflecting mutations from SRNS patients, mitigated this phenotype. We furthermore found that CRISPR/Cas9 knockout of NUP107, NUP85, or NUP133 in podocytes activated Cdc42, an important effector of SRNS pathogenesis. CRISPR/Cas9 knockout of nup107 or nup85 in zebrafish caused developmental anomalies and early lethality. In contrast, an in-frame mutation of nup107 did not affect survival, thus mimicking the allelic effects seen in humans. In conclusion, we discovered here that mutations in 4 genes encoding components of the outer ring subunits of the NPC cause SRNS and thereby provide further evidence that specific hypomorphic mutations in these essential genes cause a distinct, organ-specific phenotype.
Keywords: Genetics; Monogenic diseases; Nephrology.
Conflict of interest statement
Figures





Comment in
-
NPC mutations cause SRNS.Nat Rev Nephrol. 2018 Dec;14(12):720. doi: 10.1038/s41581-018-0064-9. Nat Rev Nephrol. 2018. PMID: 30250309 No abstract available.
-
NUP Nephropathy: When Defective Pores Cause Leaky Glomeruli.Am J Kidney Dis. 2019 Jun;73(6):890-892. doi: 10.1053/j.ajkd.2019.01.015. Epub 2019 Mar 12. Am J Kidney Dis. 2019. PMID: 30876747 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
