

Linked read technology for assembling large complex and polyploid genomes
- PMID: 30180802
- PMCID: PMC6122573
- DOI: 10.1186/s12864-018-5040-z
Linked read technology for assembling large complex and polyploid genomes
Abstract
Background: Short read DNA sequencing technologies have revolutionized genome assembly by providing high accuracy and throughput data at low cost. But it remains challenging to assemble short read data, particularly for large, complex and polyploid genomes. The linked read strategy has the potential to enhance the value of short reads for genome assembly because all reads originating from a single long molecule of DNA share a common barcode. However, the majority of studies to date that have employed linked reads were focused on human haplotype phasing and genome assembly.
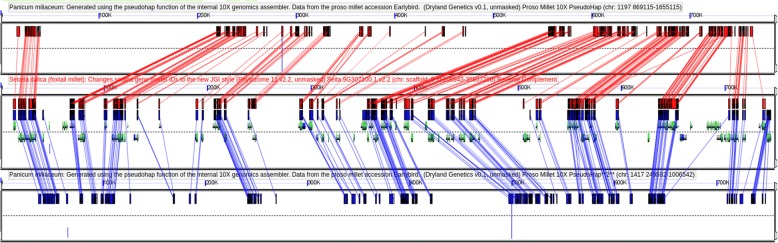
Results: Here we describe a de novo maize B73 genome assembly generated via linked read technology which contains ~ 172,000 scaffolds with an N50 of 89 kb that cover 50% of the genome. Based on comparisons to the B73 reference genome, 91% of linked read contigs are accurately assembled. Because it was possible to identify errors with > 76% accuracy using machine learning, it may be possible to identify and potentially correct systematic errors. Complex polyploids represent one of the last grand challenges in genome assembly. Linked read technology was able to successfully resolve the two subgenomes of the recent allopolyploid, proso millet (Panicum miliaceum). Our assembly covers ~ 83% of the 1 Gb genome and consists of 30,819 scaffolds with an N50 of 912 kb.
Conclusions: Our analysis provides a framework for future de novo genome assemblies using linked reads, and we suggest computational strategies that if implemented have the potential to further improve linked read assemblies, particularly for repetitive genomes.
Keywords: Genome assembly; Long molecule sequencing; Polyploid assembly.
Conflict of interest statement
Ethics approval and consent to participate
The maize B73 stock used in this study is derived from that originally obtained from Don Robertson (Iowa State University) as Schnable Lab Ac # 660; the B73 inbred is available from USDA’s National Plant Germplasm service as PI 550473. The Huntsman proso millet variety is available from the USDA NPGS as PI 578074. No permission was necessary to collect the plant samples and no specimens were deposited as vouchers.
Consent for publication
Not applicable.
Competing interests
J.C.S, C.-T.Y., and P.S.S. have equity interests in Data2Bio, LLC and Dryland Genetics, LLC.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures


References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
