

Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients
- PMID: 30180870
- PMCID: PMC6122731
- DOI: 10.1186/s13023-018-0896-1
Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients
Abstract
Background: Epidermolysis bullosa acquisita (EBA) is an orphan autoimmune disease. Several clinical phenotypes have been described, but subepidermal blistering is characteristic of all variants. Limited data on clinical and immunopathological characteristics and treatment outcomes in EBA are available. To fill this gap, we collected this information from EBA cases, meeting current diagnostic criteria, published between 1971 and 2016.
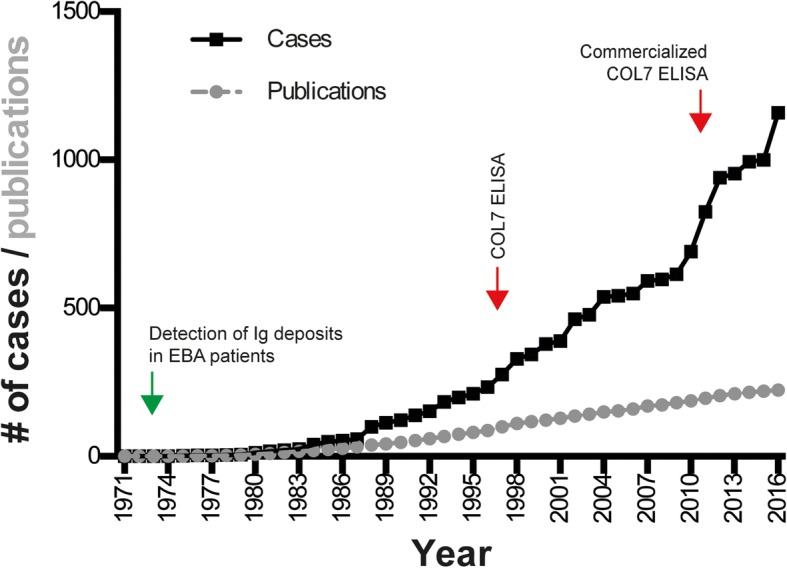
Results: We identified 1159 EBA cases. This number must be, however, interpreted with caution, as it is not possible to check for multiple reporting. The analysis of all cases indicated that EBA affects all age groups (median: 50 years, range: 1 to 94 years) at an equal gender distribution. Non-mechanobullous (non-MB) forms of EBA were observed in 55% of patients, whereas the mechanobullous variant (MB-EBA) or a combination of both variants was described in 38 or 7% of patients, respectively. Type VII collagen (COL7)-specific autoantibodies were primarily of the IgG isotype, but anti-COL7 IgA, IgM and IgE were also documented. Comparison of the 2 clinical EBA types showed a higher frequency of IgA deposits in non-MB EBA as opposed to MB EBA. Mucous membrane involvement was observed in 23% of patients, and 4.4% of cases were associated with other chronic inflammatory diseases. Of note, IgA deposits were more frequently observed in cases with mucous membrane involvement. Our analysis indicated that EBA is difficult to treat and that the choice of treatment varies widely. Chi square was applied to identify medications associated with complete remission (CR). Considering all EBA cases, intravenous immunoglobulin (IVIG, p = 0.0047) and rituximab (p = 0.0114) were associated with CR. Subgroup analysis demonstrated that no treatment was associated with CR for non-MB EBA, while IVIG (p = 0.003) was associated with CR in MB EBA.
Conclusions: Within the limitations of the study, we here document the clinical and immunopathological characteristics and treatment outcomes in a large cohort of EBA patients. The observed associations of single drugs with treatment outcome may serve as a guide to develop clinical trials.
Keywords: Diagnosis; Epidermolysis bullosa acquisita; IVIG; Meta-analysis; Rituximab; Treatment.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and approved the final version of the manuscript for publication.
Competing interests
RJL and DZ received honoraria and an unlimited research grant from Biotest. The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Elliott GT. Two cases of epidermolysis bullosa. J Cutan Genitourin Dis. 1895;13:10.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
