

Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries
- PMID: 30180884
- PMCID: PMC6124018
- DOI: 10.1186/s13059-018-1497-y
Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries
Abstract
Background: Hybridization is an important evolutionary process that results in increased plant diversity. Flowering Prunus includes popular cherry species that are appreciated worldwide for their flowers. The ornamental characteristics were acquired both naturally and through artificially hybridizing species with heterozygous genomes. Therefore, the genome of hybrid flowering Prunus presents important challenges both in plant genomics and evolutionary biology.
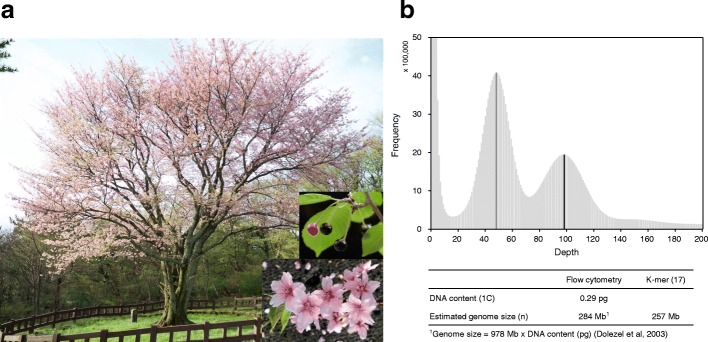
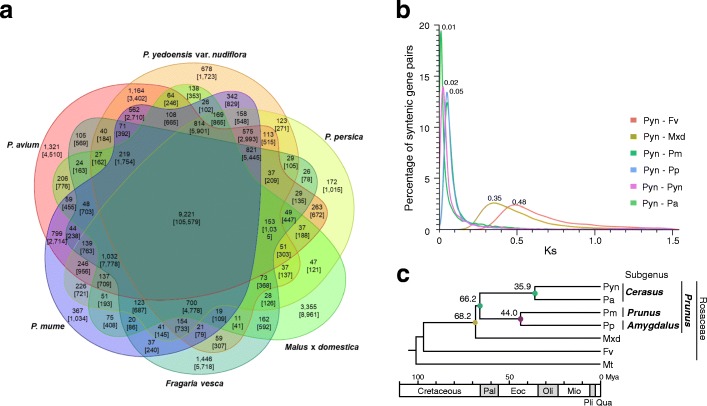
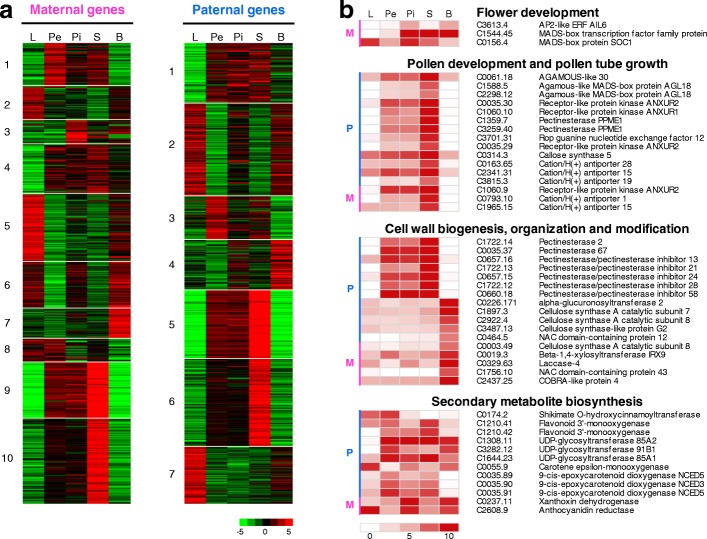
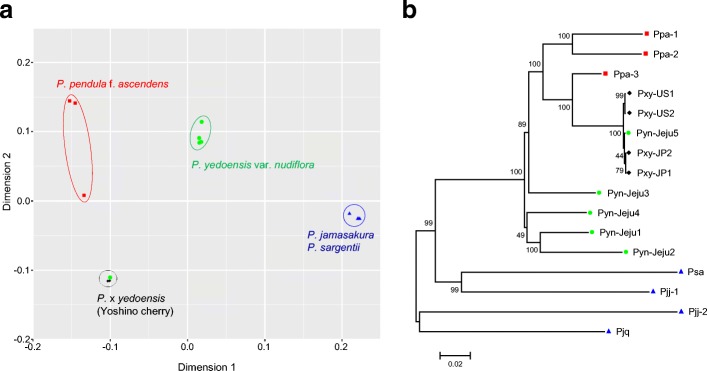
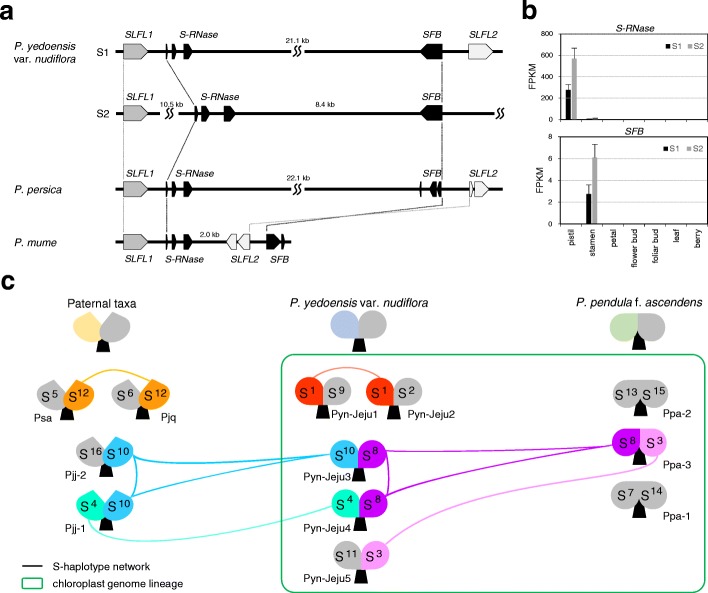
Results: We use long reads to sequence and analyze the highly heterozygous genome of wild Prunus yedoensis. The genome assembly covers > 93% of the gene space; annotation identified 41,294 protein-coding genes. Comparative analysis of the genome with 16 accessions of six related taxa shows that 41% of the genes were assigned into the maternal or paternal state. This indicates that wild P. yedoensis is an F1 hybrid originating from a cross between maternal P. pendula f. ascendens and paternal P. jamasakura, and it can be clearly distinguished from its confusing taxon, Yoshino cherry. A focused analysis of the S-locus haplotypes of closely related taxa distributed in a sympatric natural habitat suggests that reduced restriction of inter-specific hybridization due to strong gametophytic self-incompatibility is likely to promote complex hybridization of wild Prunus species and the development of a hybrid swarm.
Conclusions: We report the draft genome assembly of a natural hybrid Prunus species using long-read sequencing and sequence phasing. Based on a comprehensive comparative genome analysis with related taxa, it appears that cross-species hybridization in sympatric habitats is an ongoing process that facilitates the diversification of flowering Prunus.
Keywords: Flowering Prunus; Hybrid genome; Long-read sequencing; S-locus haplotype; Sequence phase.
Conflict of interest statement
Ethics approval and consent to participate
Wild tree samples were provided from the Korea National Arboretum. In addition, the authors obtained permission for plant sampling from Korea National Arboretum.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
