

Effects of Acetylation and Phosphorylation on Subunit Interactions in Three Large Eukaryotic Complexes
- PMID: 30181345
- PMCID: PMC6283292
- DOI: 10.1074/mcp.RA118.000892
Effects of Acetylation and Phosphorylation on Subunit Interactions in Three Large Eukaryotic Complexes
Abstract
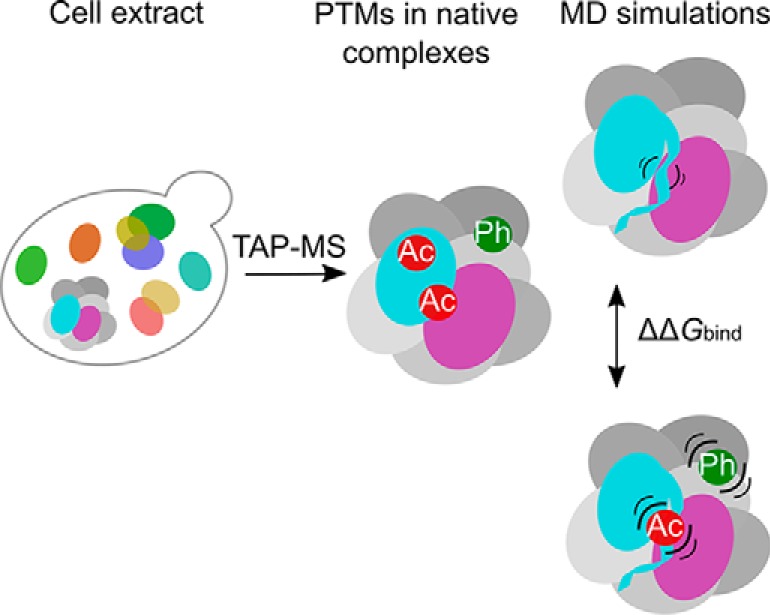
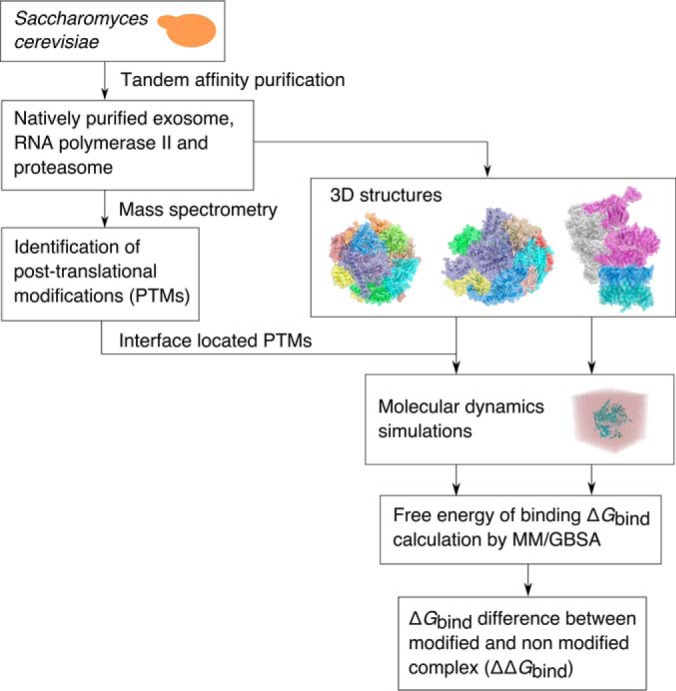
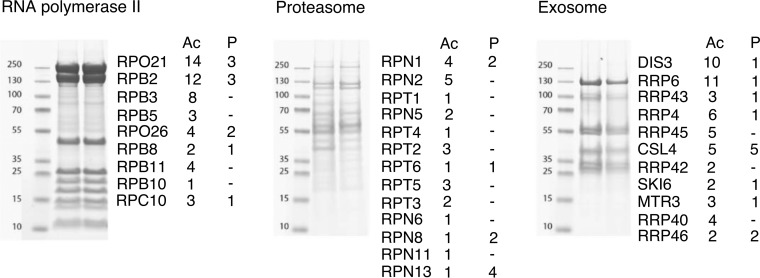
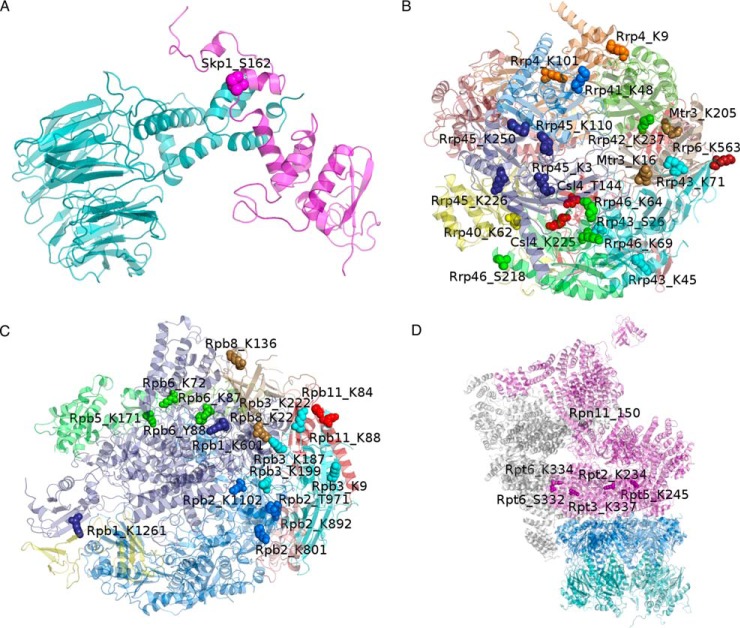
Protein post-translational modifications (PTMs) have an indispensable role in living cells as they expand chemical diversity of the proteome, providing a fine regulatory layer that can govern protein-protein interactions in changing environmental conditions. Here we investigated the effects of acetylation and phosphorylation on the stability of subunit interactions in purified Saccharomyces cerevisiae complexes, namely exosome, RNA polymerase II and proteasome. We propose a computational framework that consists of conformational sampling of the complexes by molecular dynamics simulations, followed by Gibbs energy calculation by MM/GBSA. After benchmarking against published tools such as FoldX and Mechismo, we could apply the framework for the first time on large protein assemblies with the aim of predicting the effects of PTMs located on interfaces of subunits on binding stability. We discovered that acetylation predominantly contributes to subunits' interactions in a locally stabilizing manner, while phosphorylation shows the opposite effect. Even though the local binding contributions of PTMs may be predictable to an extent, the long range effects and overall impact on subunits' binding were only captured because of our dynamical approach. Employing the developed, widely applicable workflow on other large systems will shed more light on the roles of PTMs in protein complex formation.
Keywords: Acetylation; Binding Affinity; Computational Biology; Exosome; Phosphorylation; Proteasome; Proteomics; RNA polymerase II; Structural Biology; Yeast.
© 2018 Šoštarić et al.
Figures
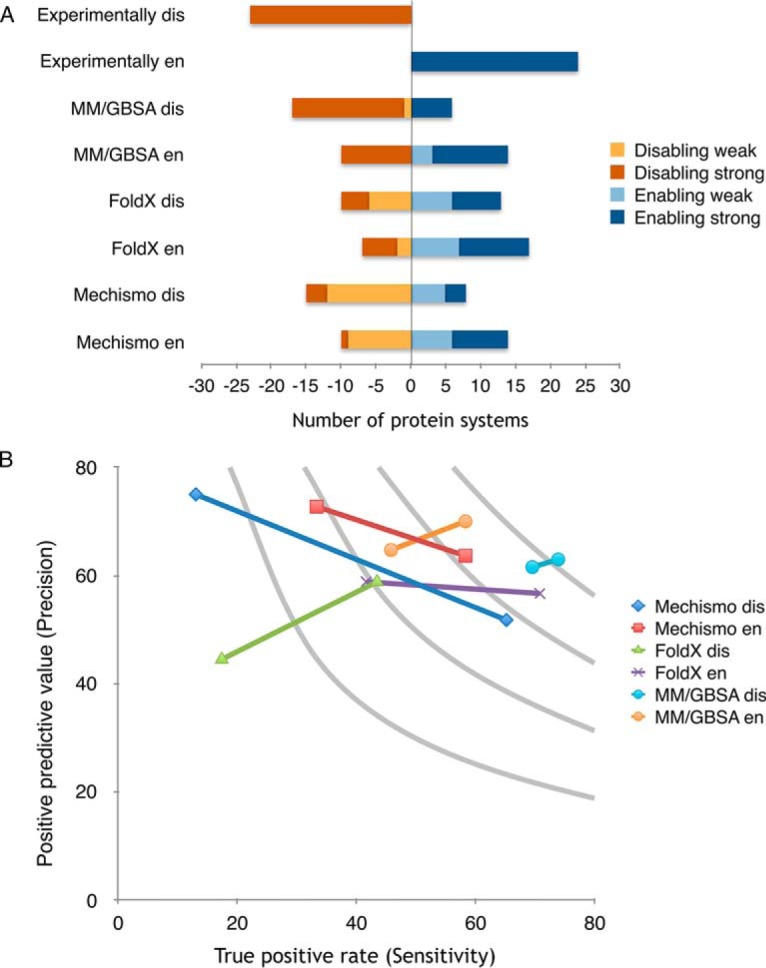
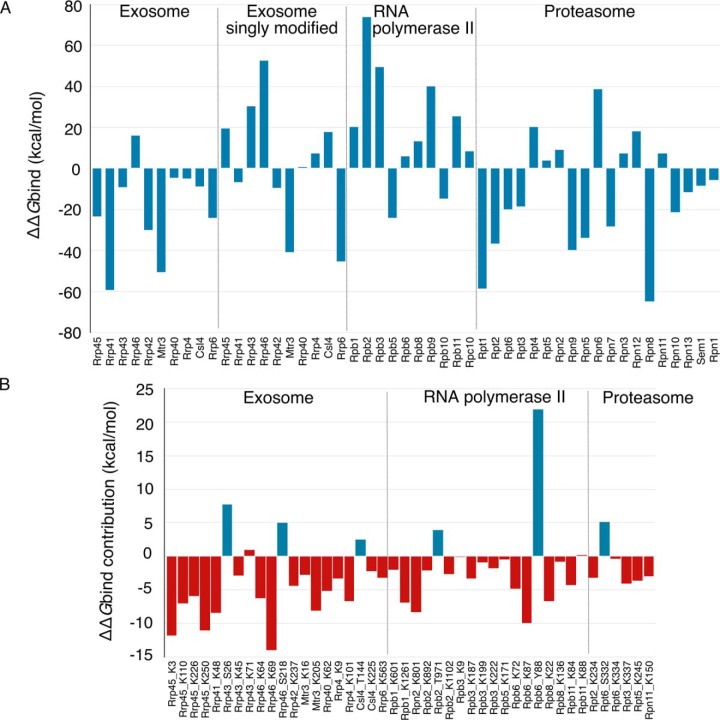
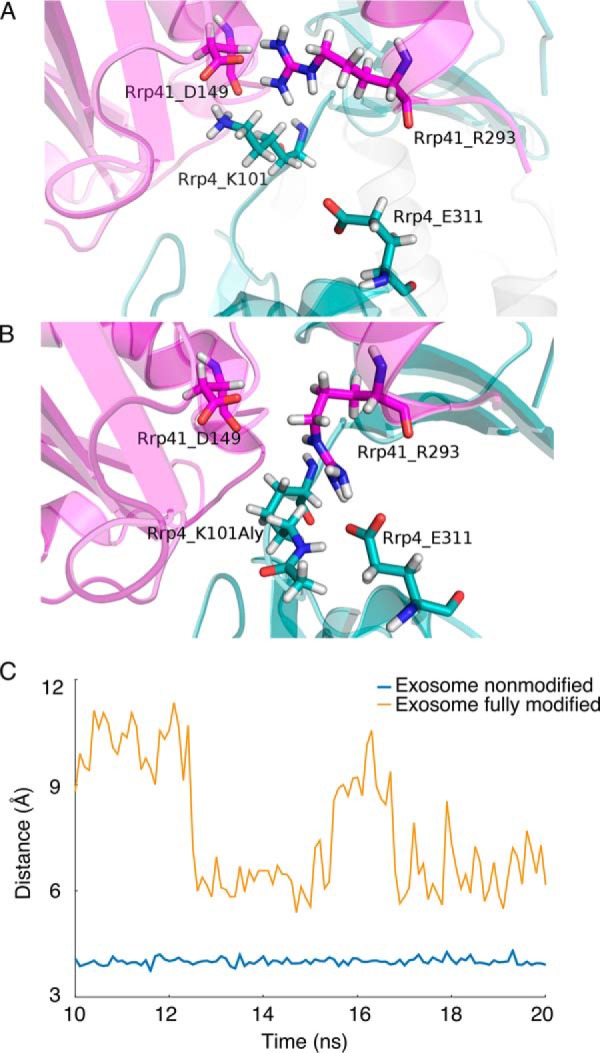
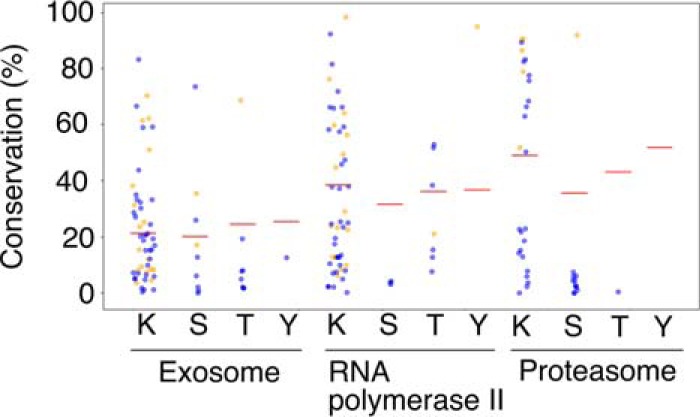
References
-
- Seet B. T., Dikic I., Zhou M.-M., and Pawson T. (2006) Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 7, 473–483 - PubMed
-
- Benayoun B. A., and Veitia R. A. (2009) A post-translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol. 19, 189–197 - PubMed
-
- Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X.-J., and Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 - PubMed
-
- Cohen P. (2001) The role of protein phosphorylation in human health and disease. Eur. J. Biochem. 268, 5001–5010 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
