

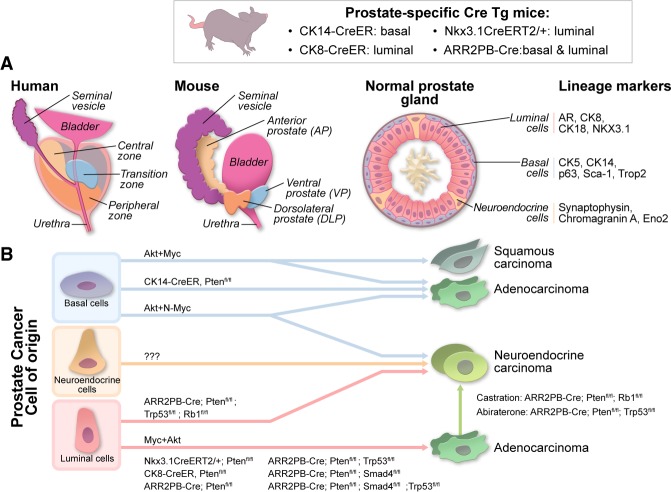
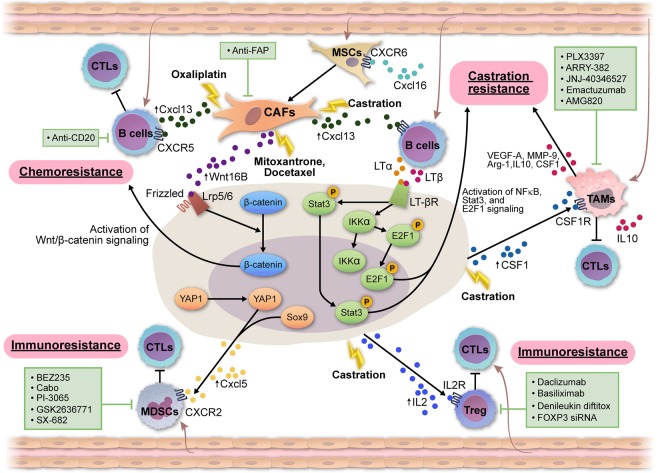
Genetics and biology of prostate cancer
- PMID: 30181359
- PMCID: PMC6120714
- DOI: 10.1101/gad.315739.118
Genetics and biology of prostate cancer
Abstract
Despite the high long-term survival in localized prostate cancer, metastatic prostate cancer remains largely incurable even after intensive multimodal therapy. The lethality of advanced disease is driven by the lack of therapeutic regimens capable of generating durable responses in the setting of extreme tumor heterogeneity on the genetic and cell biological levels. Here, we review available prostate cancer model systems, the prostate cancer genome atlas, cellular and functional heterogeneity in the tumor microenvironment, tumor-intrinsic and tumor-extrinsic mechanisms underlying therapeutic resistance, and technological advances focused on disease detection and management. These advances, along with an improved understanding of the adaptive responses to conventional cancer therapies, anti-androgen therapy, and immunotherapy, are catalyzing development of more effective therapeutic strategies for advanced disease. In particular, knowledge of the heterotypic interactions between and coevolution of cancer and host cells in the tumor microenvironment has illuminated novel therapeutic combinations with a strong potential for more durable therapeutic responses and eventual cures for advanced disease. Improved disease management will also benefit from artificial intelligence-based expert decision support systems for proper standard of care, prognostic determinant biomarkers to minimize overtreatment of localized disease, and new standards of care accelerated by next-generation adaptive clinical trials.
Keywords: prostate cancer; therapy resistance; tumor microenvironment.
© 2018 Wang et al.; Published by Cold Spring Harbor Laboratory Press.
Figures

References
-
- Aceto N, Toner M, Maheswaran S, Haber DA. 2015. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer 1: 44–52. - PubMed
-
- Alix-Panabieres C, Pantel K. 2014. Challenges in circulating tumour cell research. Nat Rev Cancer 14: 623–631. - PubMed
-
- Alix-Panabieres C, Schwarzenbach H, Pantel K. 2012. Circulating tumor cells and circulating tumor DNA. Annu Rev Med 63: 199–215. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases