

MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification
- PMID: 30181563
- PMCID: PMC6720105
- DOI: 10.1038/s41379-018-0120-9
MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification
Abstract
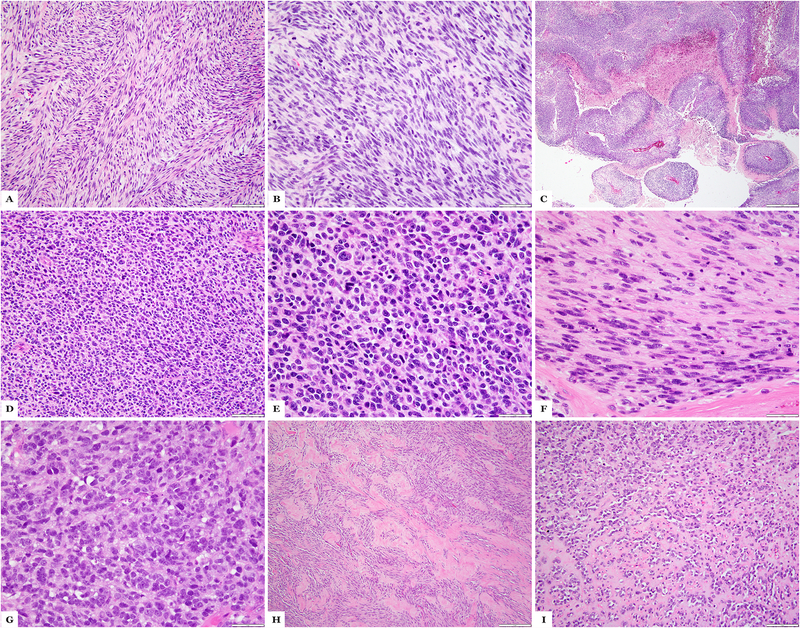
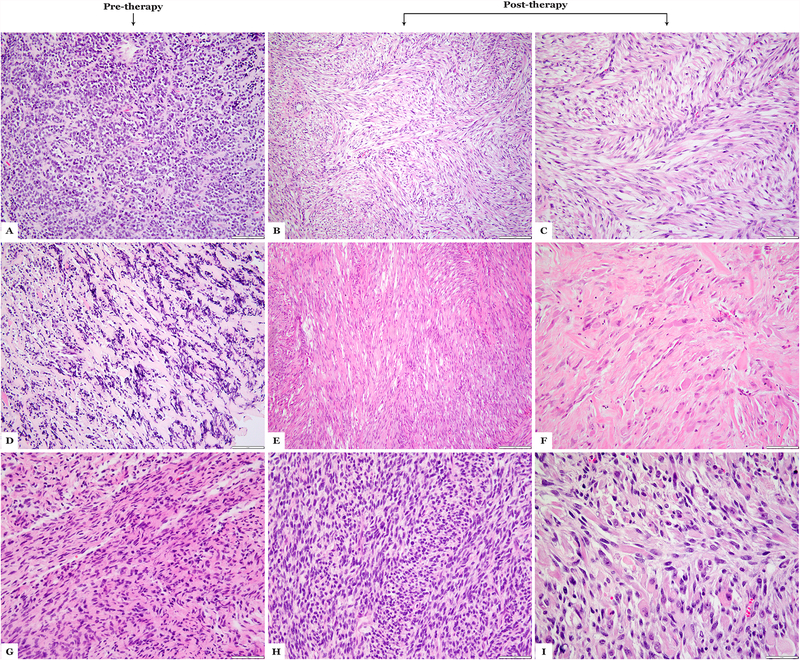
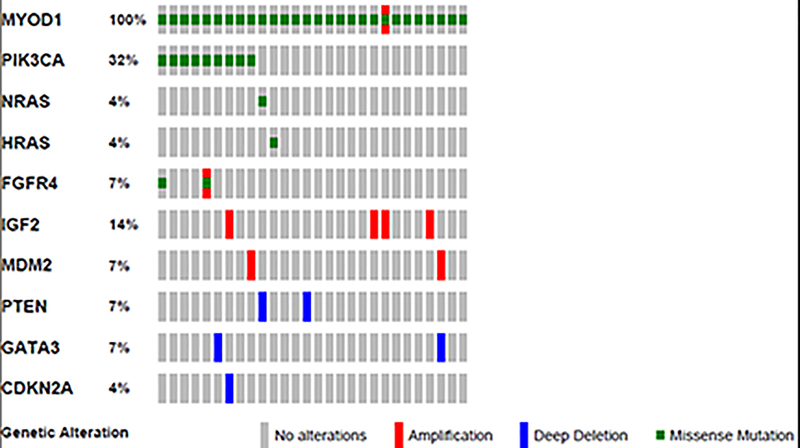
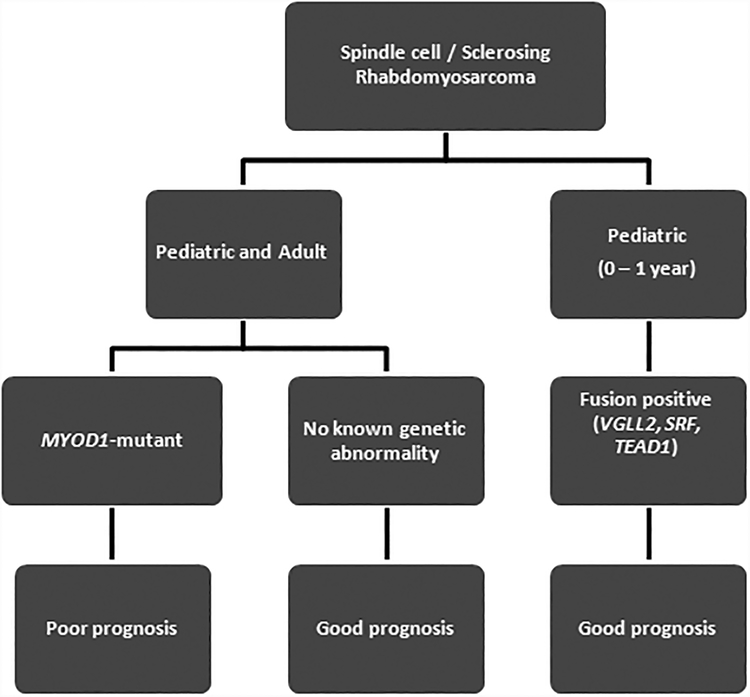
Sclerosing and spindle cell rhabdomyosarcoma is a rare histologic subtype, designated in the latest WHO classification as a stand-alone pathologic entity. Three genomic groups have been defined: an infantile subset of spindle cell rhabdomyosarcoma harboring VGLL2-related gene fusions, a MYOD1-mutant subset commonly associated with sclerosing morphology, and a subset lacking recurrent genetic abnormalities. In this study, we focus on MYOD1-mutant rhabdomyosarcoma to further define their clinicopathologic characteristics and behavior in a larger patient cohort. We investigated 30 cases of MYOD1-mutant rhabdomyosarcoma (12 previously reported and 18 newly diagnosed) with an age range of 2-94 years, including 15 children. All cases showed morphology within the spectrum of spindle cell/sclerosing rhabdomyosarcoma (8 cases showing pure sclerosing morphology, 8 cases showing pure spindle cell morphology and 14 cases showing a hybrid phenotype of spindle, sclerosing and primitive undifferentiated areas). All tumors harbored either homozygous or heterozygous MYOD1 (p.L122R) exon 1 mutations. In 10 (33%) cases, a co-existent PIK3CA mutation was identified. Hot-spot mutations in NRAS and HRAS were each identified in a single case, respectively. Follow-up was available on 22 (73%) patients with a median duration of 28 months. Local recurrence was seen in 12 (55%) and distant recurrence in 12 (55%) cases, despite multimodality chemoradiation therapy. At last follow-up, 15 (68%) patients died of the disease, one patient was alive with disease and five had no evidence of disease. The prognosis was equally poor in pediatric and adult patients. In conclusion, MYOD1 mutation defines an aggressive rhabdomyosarcoma subset, with poor outcome and response to therapy, irrespective of age. Given that this distinct molecular subtype is characterized by an aggressive biologic behavior compared to other genetic subtypes of spindle and sclerosing rhabdomyosarcoma, the MYOD1 genotype should be used as a molecular marker in both subclassification and prognostication of rhabdomyosarcoma.
Figures

References
-
- Fletcher CD, Bridge JA, Hogendoorn PC, et al. WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC press, 2013: Ch 7; p.127–35.
-
- Cavazzana AO, Schmidt D, Ninfo V, et al. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol. 1992;16:229–35. - PubMed
-
- Leuschner I, Newton WA Jr., Schmidt D, et al. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol. 1993;17:221–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P50 CA 140146-01/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)/International
- P30 CA008748/CA/NCI NIH HHS/United States
- P50 CA217694/CA/NCI NIH HHS/United States
- P01 CA047179/CA/NCI NIH HHS/United States
- PO1 CA047179-15A2/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
