

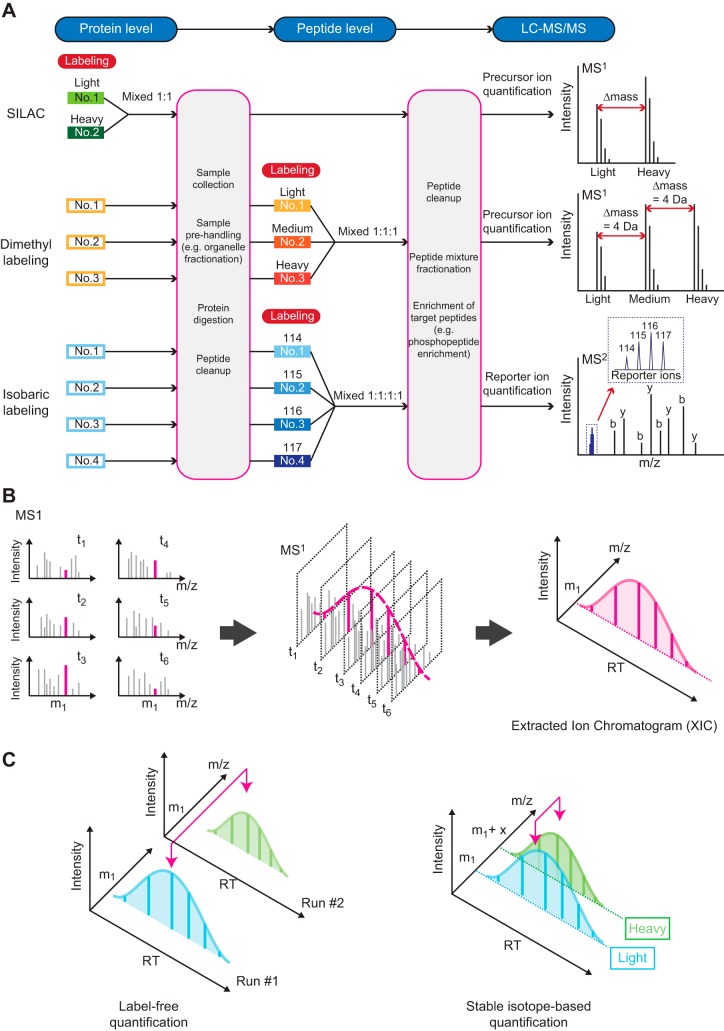
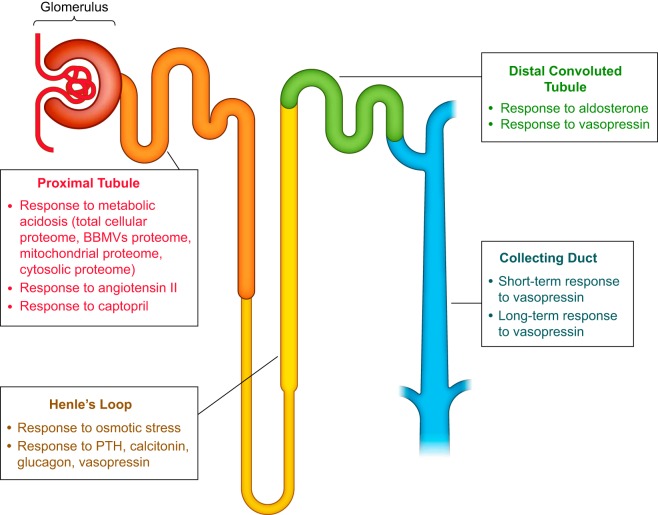
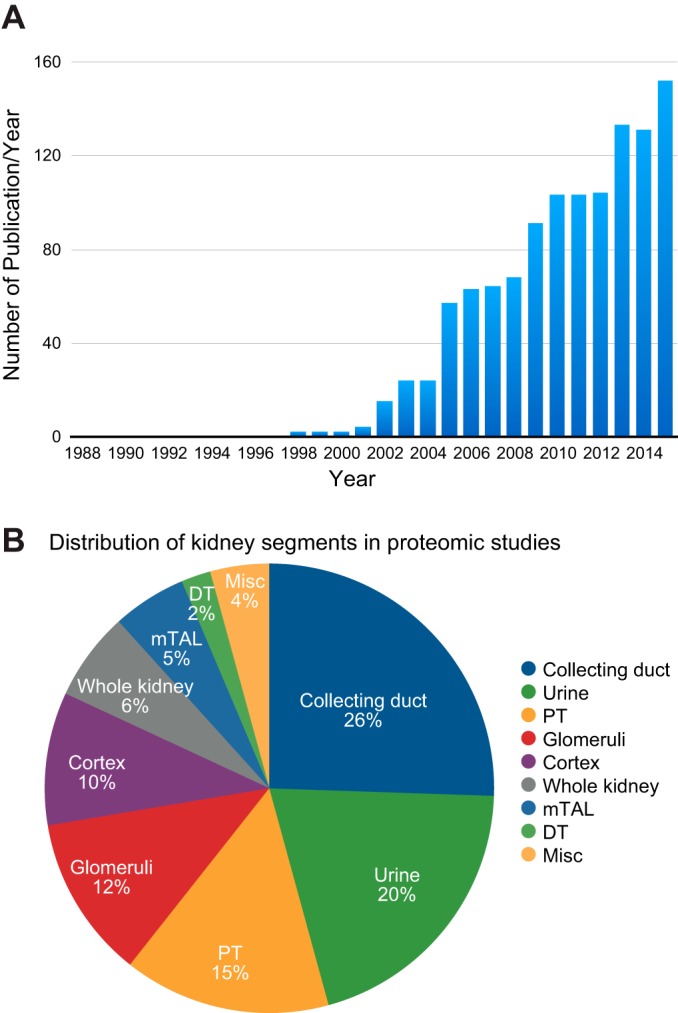
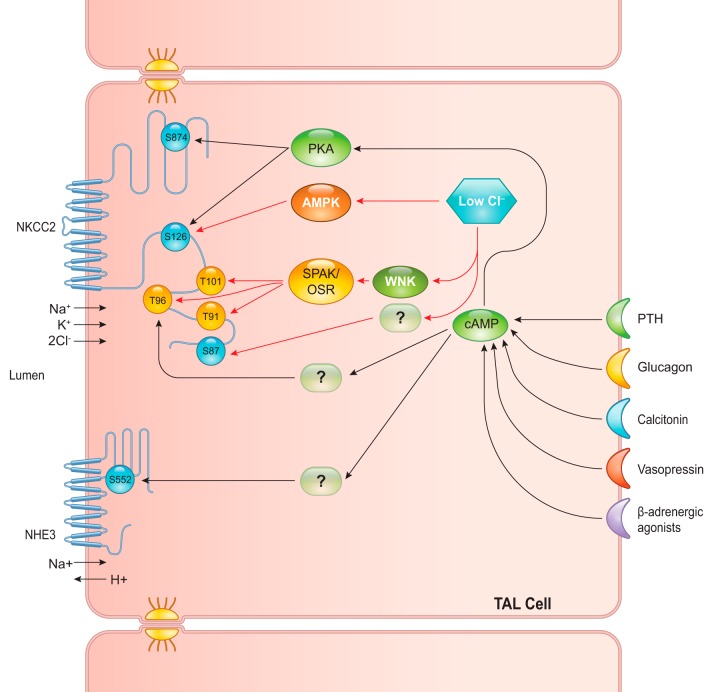
From Molecules to Mechanisms: Functional Proteomics and Its Application to Renal Tubule Physiology
- PMID: 30182799
- PMCID: PMC6335097
- DOI: 10.1152/physrev.00057.2017
From Molecules to Mechanisms: Functional Proteomics and Its Application to Renal Tubule Physiology
Abstract
Classical physiological studies using electrophysiological, biophysical, biochemical, and molecular techniques have created a detailed picture of molecular transport, bioenergetics, contractility and movement, and growth, as well as the regulation of these processes by external stimuli in cells and organisms. Newer systems biology approaches are beginning to provide deeper and broader understanding of these complex biological processes and their dynamic responses to a variety of environmental cues. In the past decade, advances in mass spectrometry-based proteomic technologies have provided invaluable tools to further elucidate these complex cellular processes, thereby confirming, complementing, and advancing common views of physiology. As one notable example, the application of proteomics to study the regulation of kidney function has yielded novel insights into the chemical and physical processes that tightly control body fluids, electrolytes, and metabolites to provide optimal microenvironments for various cellular and organ functions. Here, we systematically review, summarize, and discuss the most significant key findings from functional proteomic studies in renal epithelial physiology. We also identify further improvements in technological and bioinformatics methods that will be essential to advance precision medicine in nephrology.
Figures
Similar articles
-
Single-platform 'multi-omic' profiling: unified mass spectrometry and computational workflows for integrative proteomics-metabolomics analysis.Mol Omics. 2018 Oct 8;14(5):307-319. doi: 10.1039/c8mo00136g. Mol Omics. 2018. PMID: 30211418 Review.
-
Bioinformatics Resources for Interpreting Proteomics Mass Spectrometry Data.Methods Mol Biol. 2017;1647:267-295. doi: 10.1007/978-1-4939-7201-2_19. Methods Mol Biol. 2017. PMID: 28809010
-
Proteomics and tubulopathies.J Nephrol. 2010 Nov-Dec;23 Suppl 16:S221-7. J Nephrol. 2010. PMID: 21170884 Review.
-
Quantification of molecular heterogeneity in kidney tissue by targeted proteomics.J Proteomics. 2019 Feb 20;193:85-92. doi: 10.1016/j.jprot.2018.03.001. Epub 2018 Mar 6. J Proteomics. 2019. PMID: 29522878 Review.
-
Quantitative Proteomics of All 14 Renal Tubule Segments in Rat.J Am Soc Nephrol. 2020 Jun;31(6):1255-1266. doi: 10.1681/ASN.2020010071. Epub 2020 May 1. J Am Soc Nephrol. 2020. PMID: 32358040 Free PMC article.
Cited by
-
Advances in Diagnosis and Treatment of Inherited Kidney Diseases in Children.Kidney Dis (Basel). 2024 Sep 24;10(6):558-572. doi: 10.1159/000541564. eCollection 2024 Dec. Kidney Dis (Basel). 2024. PMID: 39664340 Free PMC article. Review.
-
Proteomics and AQP2 regulation.J Physiol. 2024 Jul;602(13):3011-3023. doi: 10.1113/JP283899. Epub 2023 Jan 12. J Physiol. 2024. PMID: 36571566 Free PMC article. Review.
-
H3 relaxin protects against calcium oxalate crystal-induced renal inflammatory pyroptosis.Cell Prolif. 2020 Oct;53(10):e12902. doi: 10.1111/cpr.12902. Epub 2020 Sep 18. Cell Prolif. 2020. PMID: 32945585 Free PMC article.
-
Proteomics and Extracellular Vesicles as Novel Biomarker Sources in Peritoneal Dialysis in Children.Int J Mol Sci. 2022 May 18;23(10):5655. doi: 10.3390/ijms23105655. Int J Mol Sci. 2022. PMID: 35628461 Free PMC article. Review.
-
OMICS in Chronic Kidney Disease: Focus on Prognosis and Prediction.Int J Mol Sci. 2021 Dec 29;23(1):336. doi: 10.3390/ijms23010336. Int J Mol Sci. 2021. PMID: 35008760 Free PMC article. Review.
References
-
- Abbatiello SE, Schilling B, Mani DR, Zimmerman LJ, Hall SC, MacLean B, Albertolle M, Allen S, Burgess M, Cusack MP, Gosh M, Hedrick V, Held JM, Inerowicz HD, Jackson A, Keshishian H, Kinsinger CR, Lyssand J, Makowski L, Mesri M, Rodriguez H, Rudnick P, Sadowski P, Sedransk N, Shaddox K, Skates SJ, Kuhn E, Smith D, Whiteaker JR, Whitwell C, Zhang S, Borchers CH, Fisher SJ, Gibson BW, Liebler DC, MacCoss MJ, Neubert TA, Paulovich AG, Regnier FE, Tempst P, Carr SA. Large-scale interlaboratory study to develop, analytically validate and apply highly multiplexed, quantitative peptide assays to measure cancer-relevant proteins in plasma. Mol Cell Proteomics 14: 2357–2374, 2015. doi:10.1074/mcp.M114.047050. - DOI - PMC - PubMed
-
- Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Gil L, Girón CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Juettemann T, Keenan S, Laird MR, Lavidas I, Maurel T, McLaren W, Moore B, Murphy DN, Nag R, Newman V, Nuhn M, Ong CK, Parker A, Patricio M, Riat HS, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Wilder SP, Zadissa A, Kostadima M, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Cunningham F, Yates A, Zerbino DR, Flicek P; Ensembl 2017 . Nucleic Acids Res 45: D635–D642, 2017. doi:10.1093/nar/gkw1104. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous