

SUMO-1 modification of FEN1 facilitates its interaction with Rad9-Rad1-Hus1 to counteract DNA replication stress
- PMID: 30184152
- PMCID: PMC6231531
- DOI: 10.1093/jmcb/mjy047
SUMO-1 modification of FEN1 facilitates its interaction with Rad9-Rad1-Hus1 to counteract DNA replication stress
Abstract
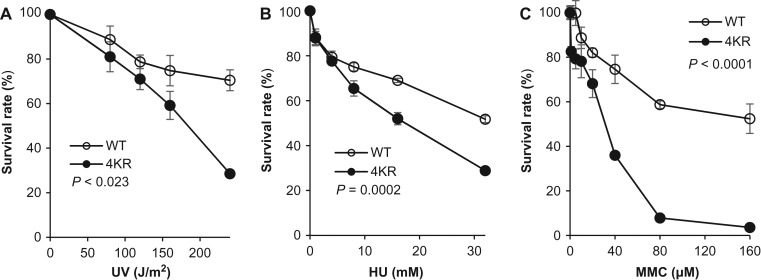
Human flap endonuclease 1 (FEN1) is a structure-specific, multi-functional endonuclease essential for DNA replication and repair. We and others have shown that during DNA replication, FEN1 processes Okazaki fragments via its interaction with the proliferating cell nuclear antigen (PCNA). Alternatively, in response to DNA damage, FEN1 interacts with the PCNA-like Rad9-Rad1-Hus1 complex instead of PCNA to engage in DNA repair activities, such as homology-directed repair of stalled DNA replication forks. However, it is unclear how FEN1 is able to switch between these interactions and its roles in DNA replication and DNA repair. Here, we report that FEN1 undergoes SUMOylation by SUMO-1 in response to DNA replication fork-stalling agents, such as UV irradiation, hydroxyurea, and mitomycin C. This DNA damage-induced SUMO-1 modification promotes the interaction of FEN1 with the Rad9-Rad1-Hus1 complex. Furthermore, we found that FEN1 mutations that prevent its SUMO-1 modification also impair its ability to interact with HUS1 and to rescue stalled replication forks. These impairments lead to the accumulation of DNA damage and heightened sensitivity to fork-stalling agents. Altogether, our findings suggest an important role of the SUMO-1 modification of FEN1 in regulating its roles in DNA replication and repair.
Figures







Similar articles
-
Succinylation at a key residue of FEN1 is involved in the DNA damage response to maintain genome stability.Am J Physiol Cell Physiol. 2020 Oct 1;319(4):C657-C666. doi: 10.1152/ajpcell.00137.2020. Epub 2020 Aug 12. Am J Physiol Cell Physiol. 2020. PMID: 32783654
-
The human Rad9-Rad1-Hus1 checkpoint complex stimulates flap endonuclease 1.Proc Natl Acad Sci U S A. 2004 Nov 30;101(48):16762-7. doi: 10.1073/pnas.0407686101. Epub 2004 Nov 19. Proc Natl Acad Sci U S A. 2004. PMID: 15556996 Free PMC article.
-
Repair complexes of FEN1 endonuclease, DNA, and Rad9-Hus1-Rad1 are distinguished from their PCNA counterparts by functionally important stability.Proc Natl Acad Sci U S A. 2012 May 29;109(22):8528-33. doi: 10.1073/pnas.1121116109. Epub 2012 May 14. Proc Natl Acad Sci U S A. 2012. PMID: 22586102 Free PMC article.
-
Evidence that DNA damage detection machinery participates in DNA repair.Cell Cycle. 2005 Apr;4(4):529-32. doi: 10.4161/cc.4.4.1598. Epub 2005 Apr 10. Cell Cycle. 2005. PMID: 15876866 Review.
-
Functional regulation of FEN1 nuclease and its link to cancer.Nucleic Acids Res. 2011 Feb;39(3):781-94. doi: 10.1093/nar/gkq884. Epub 2010 Oct 6. Nucleic Acids Res. 2011. PMID: 20929870 Free PMC article. Review.
Cited by
-
Symmetrical dimethylation of H4R3: A bridge linking DNA damage and repair upon oxidative stress.Redox Biol. 2020 Oct;37:101653. doi: 10.1016/j.redox.2020.101653. Epub 2020 Jul 24. Redox Biol. 2020. PMID: 32739156 Free PMC article.
-
Investigation of the Possible Role of RAD9 in Post-Diapaused Embryonic Development of the Brine Shrimp Artemia sinica.Genes (Basel). 2019 Sep 30;10(10):768. doi: 10.3390/genes10100768. Genes (Basel). 2019. PMID: 31574972 Free PMC article.
-
The multifaceted roles of DNA repair and replication proteins in aging and obesity.DNA Repair (Amst). 2021 Mar;99:103049. doi: 10.1016/j.dnarep.2021.103049. Epub 2021 Jan 21. DNA Repair (Amst). 2021. PMID: 33529944 Free PMC article. Review.
-
FEN1 Promotes Hepatocellular Carcinoma Progression by Activating Cell Cycle Transition from G2 To M Phase.J Cancer. 2024 Jan 1;15(4):981-989. doi: 10.7150/jca.88160. eCollection 2024. J Cancer. 2024. PMID: 38230217 Free PMC article.
-
Functional regulation of the structure-specific endonuclease FEN1 by the human cytomegalovirus protein IE1 suggests a role for the re-initiation of stalled viral replication forks.PLoS Pathog. 2021 Mar 26;17(3):e1009460. doi: 10.1371/journal.ppat.1009460. eCollection 2021 Mar. PLoS Pathog. 2021. PMID: 33770148 Free PMC article.
References
-
- Ayyagari R., Gomes X.V., Gordenin D.A., et al. . (2003). Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 and DNA2. J. Biol. Chem. 278, 1618–1625. - PubMed
-
- Bae S.H., Bae K.H., Kim J.A., et al. . (2001). RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature 412, 456–461. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
