

Genetic screen identifies adaptive aneuploidy as a key mediator of ER stress resistance in yeast
- PMID: 30185560
- PMCID: PMC6156608
- DOI: 10.1073/pnas.1804264115
Genetic screen identifies adaptive aneuploidy as a key mediator of ER stress resistance in yeast
Abstract
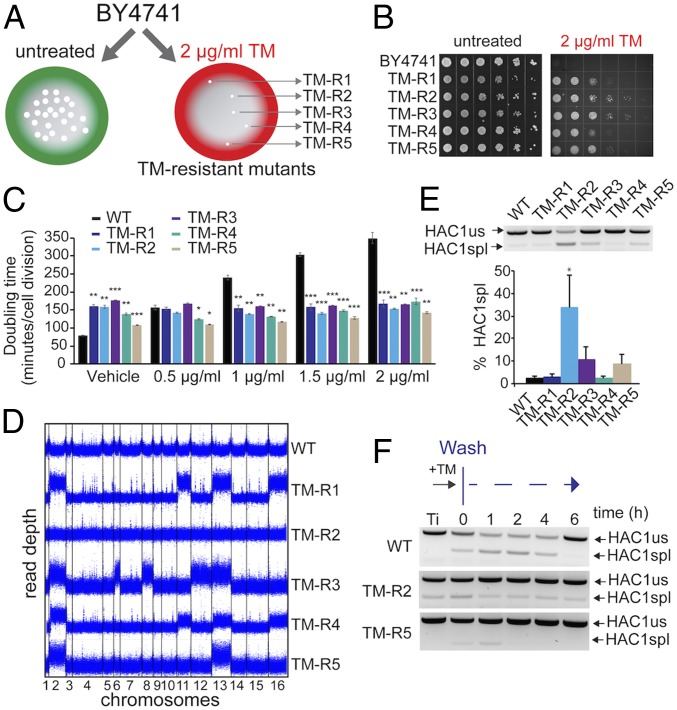
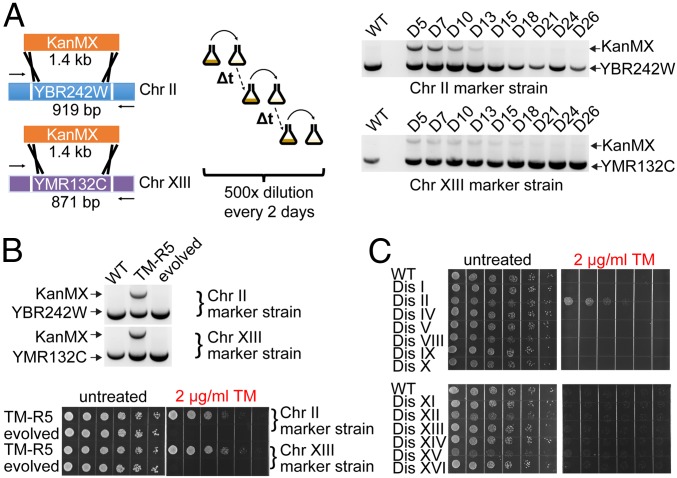
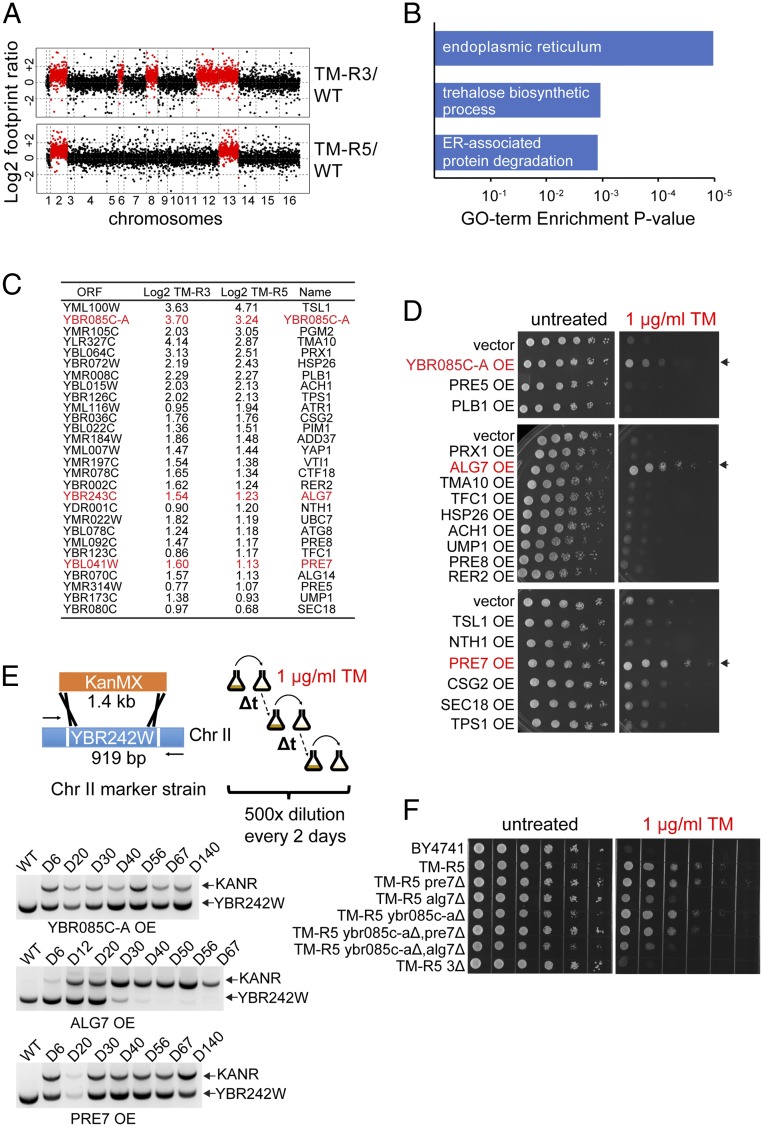
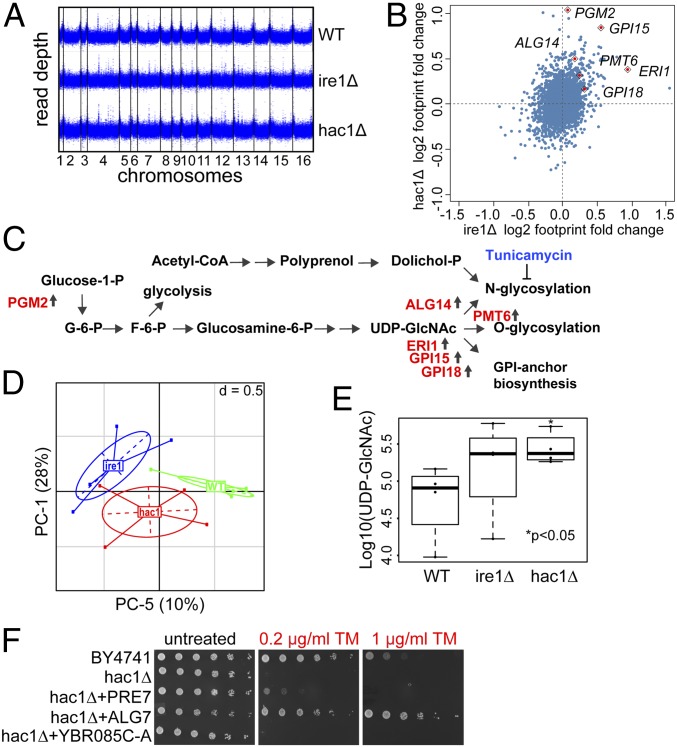
The yeast genome becomes unstable during stress, which often results in adaptive aneuploidy, allowing rapid activation of protective mechanisms that restore cellular homeostasis. In this study, we performed a genetic screen in Saccharomyces cerevisiae to identify genome adaptations that confer resistance to tunicamycin-induced endoplasmic reticulum (ER) stress. Whole-genome sequencing of tunicamycin-resistant mutants revealed that ER stress resistance correlated significantly with gains of chromosomes II and XIII. We found that chromosome duplications allow adaptation of yeast cells to ER stress independently of the unfolded protein response, and that the gain of an extra copy of chromosome II alone is sufficient to induce protection from tunicamycin. Moreover, the protective effect of disomic chromosomes can be recapitulated by overexpression of several genes located on chromosome II. Among these genes, overexpression of UDP-N-acetylglucosamine-1-P transferase (ALG7), a subunit of the 20S proteasome (PRE7), and YBR085C-A induced tunicamycin resistance in wild-type cells, whereas deletion of all three genes completely reversed the tunicamycin-resistance phenotype. Together, our data demonstrate that aneuploidy plays a critical role in adaptation to ER stress by increasing the copy number of ER stress protective genes. While aneuploidy itself leads to proteotoxic stress, the gene-specific effects of chromosome II aneuploidy counteract the negative effect resulting in improved protein folding.
Keywords: ER stress resistance; Saccharomyces cerevisiae; aneuploidy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Walter P, Ron D. The unfolded protein response: From stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
