

The Coronavirus Transmissible Gastroenteritis Virus Evades the Type I Interferon Response through IRE1α-Mediated Manipulation of the MicroRNA miR-30a-5p/SOCS1/3 Axis
- PMID: 30185587
- PMCID: PMC6206482
- DOI: 10.1128/JVI.00728-18
The Coronavirus Transmissible Gastroenteritis Virus Evades the Type I Interferon Response through IRE1α-Mediated Manipulation of the MicroRNA miR-30a-5p/SOCS1/3 Axis
Abstract
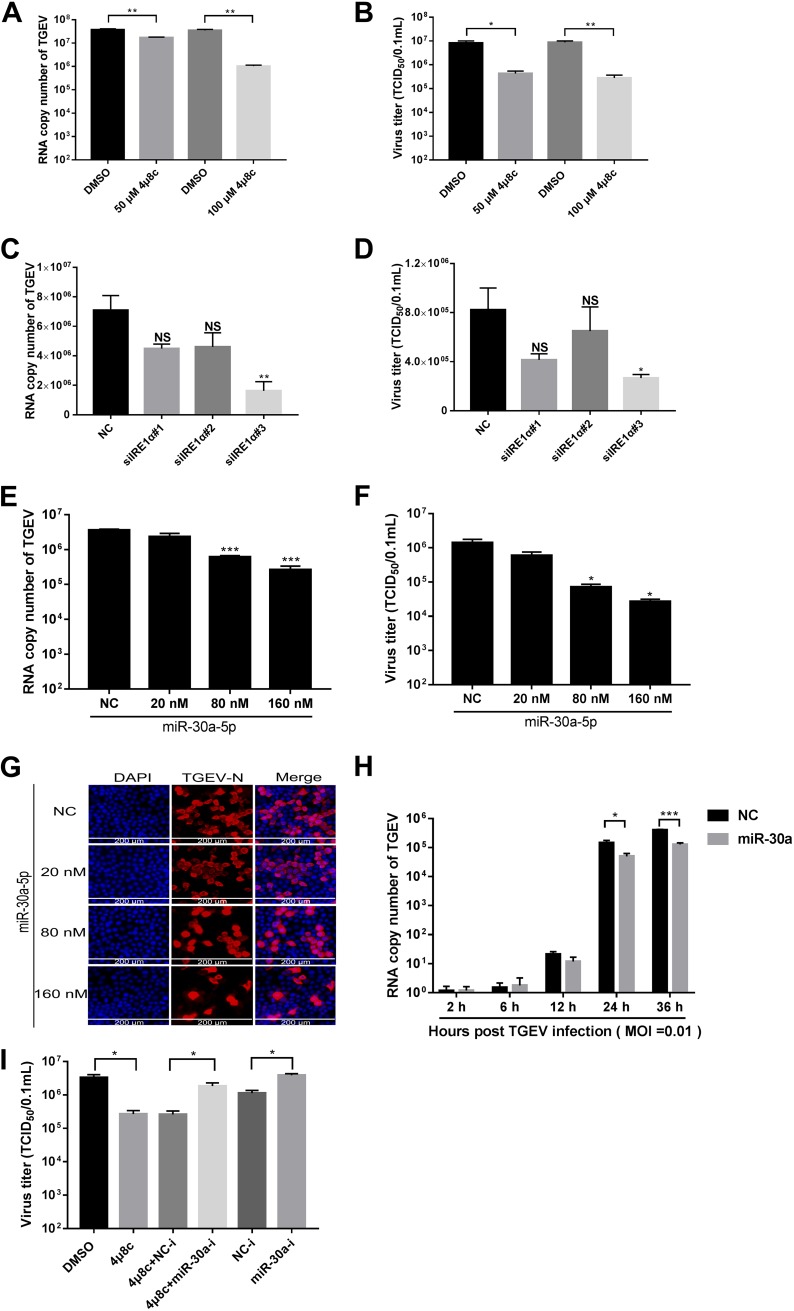
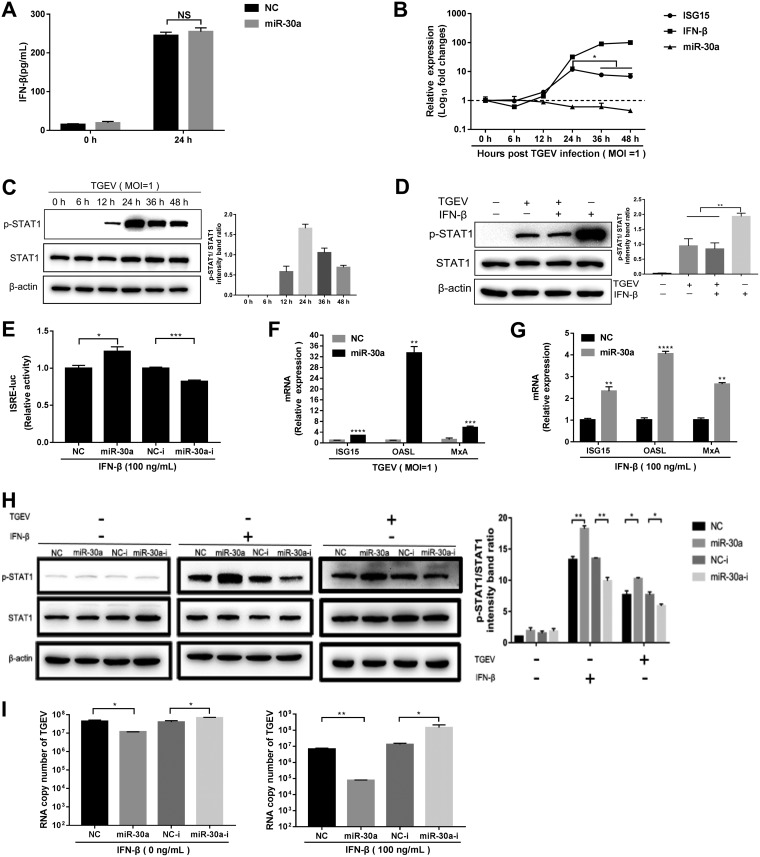
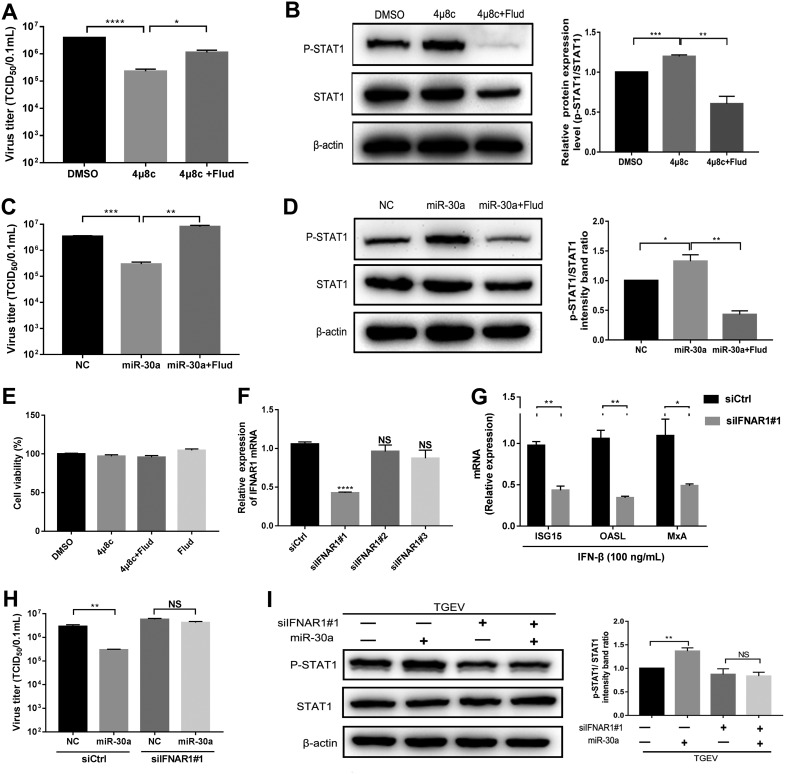
In host innate immunity, type I interferons (IFN-I) are major antiviral molecules, and coronaviruses have evolved diverse strategies to counter the IFN-I response during infection. Transmissible gastroenteritis virus (TGEV), a member of the Alphacoronavirus family, induces endoplasmic reticulum (ER) stress and significant IFN-I production after infection. However, how TGEV evades the IFN-I antiviral response despite the marked induction of endogenous IFN-I has remained unclear. Inositol-requiring enzyme 1 α (IRE1α), a highly conserved ER stress sensor with both kinase and RNase activities, is involved in the IFN response. In this study, IRE1α facilitated TGEV replication via downmodulating the host microRNA (miR) miR-30a-5p abundance. miR-30a-5p normally enhances IFN-I antiviral activity by directly targeting the negative regulators of Janus family kinase (JAK)-signal transducer and activator of transcription (STAT), the suppressor of cytokine signaling protein 1 (SOCS1), and SOCS3. Furthermore, TGEV infection increased SOCS1 and SOCS3 expression, which dampened the IFN-I antiviral response and facilitated TGEV replication. Importantly, compared with mock infection, TGEV infection in vivo resulted in decreased miR-30a-5p levels and significantly elevated SOCS1 and SOCS3 expression in the piglet ileum. Taken together, our data reveal a new strategy used by TGEV to escape the IFN-I response by engaging the IRE1α-miR-30a-5p/SOCS1/3 axis, thus improving our understanding of how TGEV escapes host innate immune defenses.IMPORTANCE Type I interferons (IFN-I) play essential roles in restricting viral infections. Coronavirus infection induces ER stress and the interferon response, which reflects different adaptive cellular processes. An understanding of how coronavirus-elicited ER stress is actively involved in viral replication and manipulates the host IFN-I response has remained elusive. Here, TGEV inhibited host miR-30a-5p via the ER stress sensor IRE1α, which led to the increased expression of negative regulators of JAK-STAT signaling cascades, namely, SOCS1 and SOCS3. Increased SOCS1 or SOCS3 expression impaired the IFN-I antiviral response, promoting TGEV replication. These findings enhance our understanding of the strategies used by coronaviruses to antagonize IFN-I innate immunity via IRE1α-mediated manipulation of the miR-30a-5p/SOCS axis, highlighting the crucial role of IRE1α in innate antiviral resistance and the potential of IRE1α as a novel target against coronavirus infection.
Keywords: IRE1α; SOCS; miR-30a-5p; transmissible gastroenteritis virus (TGEV); type I interferon.
Copyright © 2018 American Society for Microbiology.
Figures





Similar articles
-
The PERK Arm of the Unfolded Protein Response Negatively Regulates Transmissible Gastroenteritis Virus Replication by Suppressing Protein Translation and Promoting Type I Interferon Production.J Virol. 2018 Jul 17;92(15):e00431-18. doi: 10.1128/JVI.00431-18. Print 2018 Aug 1. J Virol. 2018. PMID: 29769338 Free PMC article.
-
Duck Tembusu virus promotes the expression of suppressor of cytokine signaling 1 by downregulating miR-148a-5p to facilitate virus replication.Infect Genet Evol. 2020 Nov;85:104392. doi: 10.1016/j.meegid.2020.104392. Epub 2020 Jun 10. Infect Genet Evol. 2020. PMID: 32534026
-
The Coronavirus PEDV Evades Type III Interferon Response Through the miR-30c-5p/SOCS1 Axis.Front Microbiol. 2020 May 22;11:1180. doi: 10.3389/fmicb.2020.01180. eCollection 2020. Front Microbiol. 2020. PMID: 32574254 Free PMC article.
-
Immune evasion of porcine enteric coronaviruses and viral modulation of antiviral innate signaling.Virus Res. 2016 Dec 2;226:128-141. doi: 10.1016/j.virusres.2016.05.015. Epub 2016 May 19. Virus Res. 2016. PMID: 27212682 Free PMC article. Review.
-
Regulation of the MIR155 host gene in physiological and pathological processes.Gene. 2013 Dec 10;532(1):1-12. doi: 10.1016/j.gene.2012.12.009. Epub 2012 Dec 14. Gene. 2013. PMID: 23246696 Review.
Cited by
-
Rotavirus Induces Epithelial-Mesenchymal Transition Markers by Transcriptional Suppression of miRNA-29b.Front Microbiol. 2021 Feb 18;12:631183. doi: 10.3389/fmicb.2021.631183. eCollection 2021. Front Microbiol. 2021. PMID: 33679655 Free PMC article.
-
An Overview of Current Knowledge of Deadly CoVs and Their Interface with Innate Immunity.Viruses. 2021 Mar 26;13(4):560. doi: 10.3390/v13040560. Viruses. 2021. PMID: 33810391 Free PMC article. Review.
-
Differential Transcriptomics Analysis of IPEC-J2 Cells Single or Coinfected With Porcine Epidemic Diarrhea Virus and Transmissible Gastroenteritis Virus.Front Immunol. 2022 Mar 25;13:844657. doi: 10.3389/fimmu.2022.844657. eCollection 2022. Front Immunol. 2022. PMID: 35401515 Free PMC article.
-
Megalocytivirus Induces Complicated Fish Immune Response at Multiple RNA Levels Involving mRNA, miRNA, and circRNA.Int J Mol Sci. 2021 Mar 19;22(6):3156. doi: 10.3390/ijms22063156. Int J Mol Sci. 2021. PMID: 33808870 Free PMC article.
-
The role of K63-linked polyubiquitin in several types of autophagy.Biol Futur. 2022 Jun;73(2):137-148. doi: 10.1007/s42977-022-00117-4. Epub 2022 May 2. Biol Futur. 2022. PMID: 35501575 Review.
References
-
- Favreau DJ, Desforges M, St-Jean JR, Talbot PJ. 2009. A human coronavirus OC43 variant harboring persistence-associated mutations in the S glycoprotein differentially induces the unfolded protein response in human neurons as compared to wild-type virus. Virology 395:255–267. doi:10.1016/j.virol.2009.09.026. - DOI - PMC - PubMed
-
- DeDiego ML, Nieto-Torres JL, Jimenez-Guardeno JM, Regla-Nava JA, Alvarez E, Oliveros JC, Zhao J, Fett C, Perlman S, Enjuanes L. 2011. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog 7:e1002315. doi:10.1371/journal.ppat.1002315. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
