

Combining discovery and targeted proteomics reveals a prognostic signature in oral cancer
- PMID: 30185791
- PMCID: PMC6125363
- DOI: 10.1038/s41467-018-05696-2
Combining discovery and targeted proteomics reveals a prognostic signature in oral cancer
Abstract
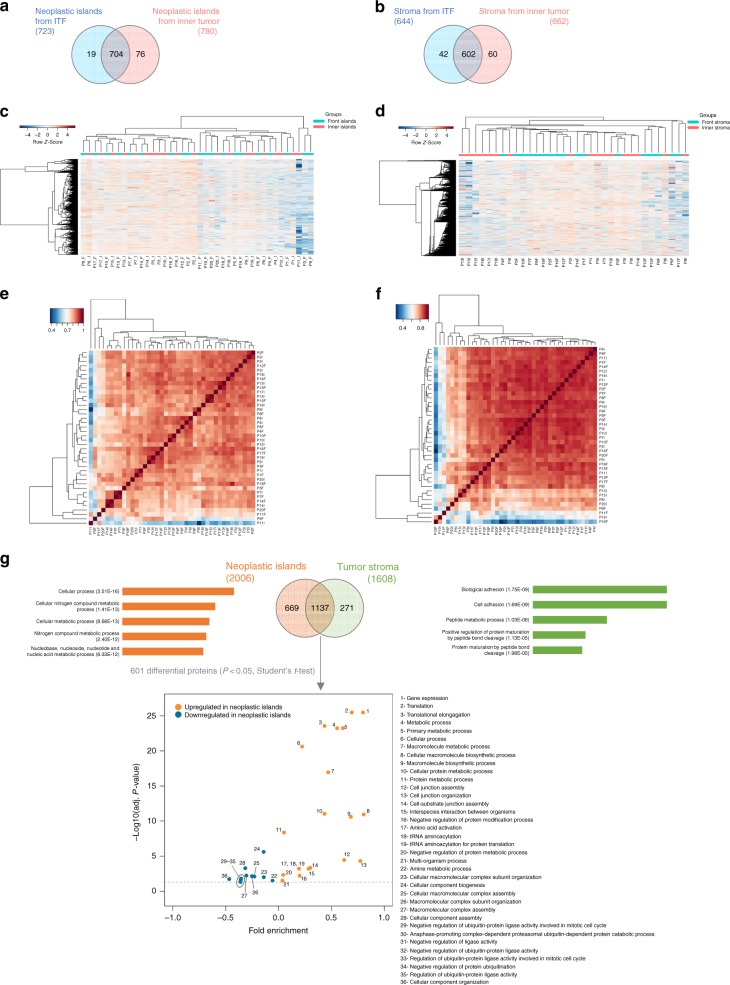
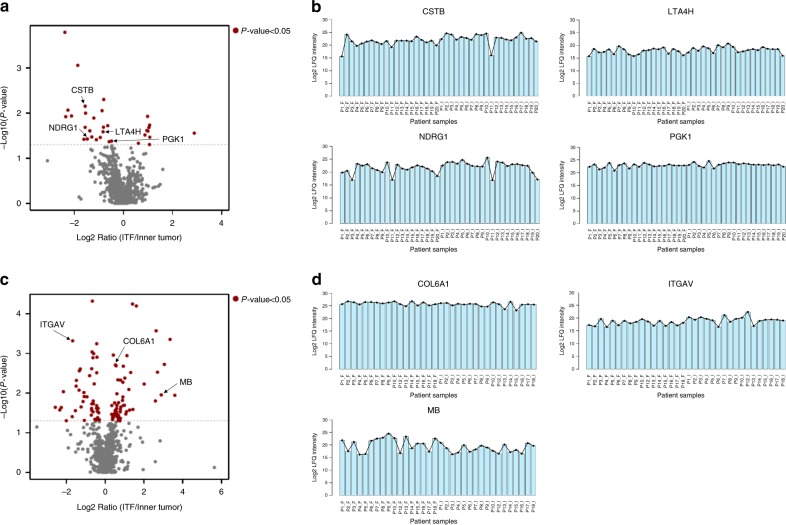
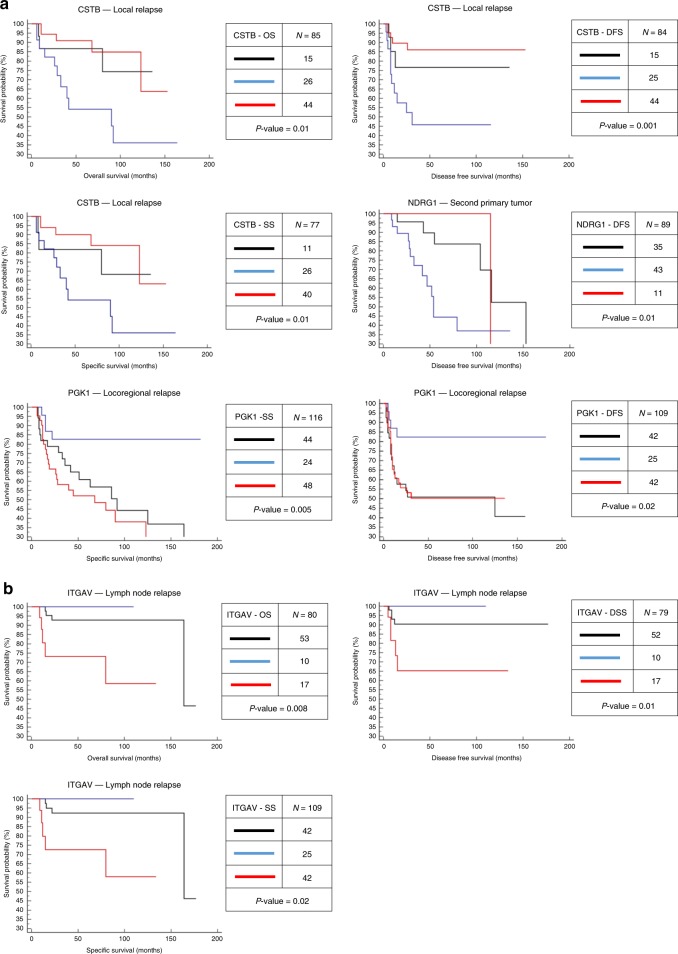
Different regions of oral squamous cell carcinoma (OSCC) have particular histopathological and molecular characteristics limiting the standard tumor-node-metastasis prognosis classification. Therefore, defining biological signatures that allow assessing the prognostic outcomes for OSCC patients would be of great clinical significance. Using histopathology-guided discovery proteomics, we analyze neoplastic islands and stroma from the invasive tumor front (ITF) and inner tumor to identify differentially expressed proteins. Potential signature proteins are prioritized and further investigated by immunohistochemistry (IHC) and targeted proteomics. IHC indicates low expression of cystatin-B in neoplastic islands from the ITF as an independent marker for local recurrence. Targeted proteomics analysis of the prioritized proteins in saliva, combined with machine-learning methods, highlights a peptide-based signature as the most powerful predictor to distinguish patients with and without lymph node metastasis. In summary, we identify a robust signature, which may enhance prognostic decisions in OSCC and better guide treatment to reduce tumor recurrence or lymph node metastasis.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
