

Extensive sex differences at the initiation of genetic recombination
- PMID: 30185906
- PMCID: PMC6364566
- DOI: 10.1038/s41586-018-0492-5
Extensive sex differences at the initiation of genetic recombination
Abstract
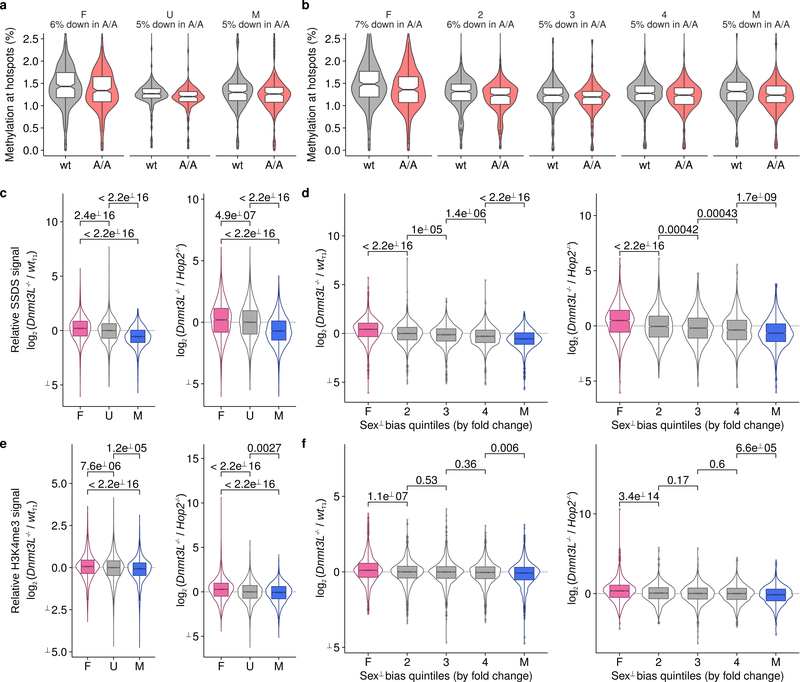
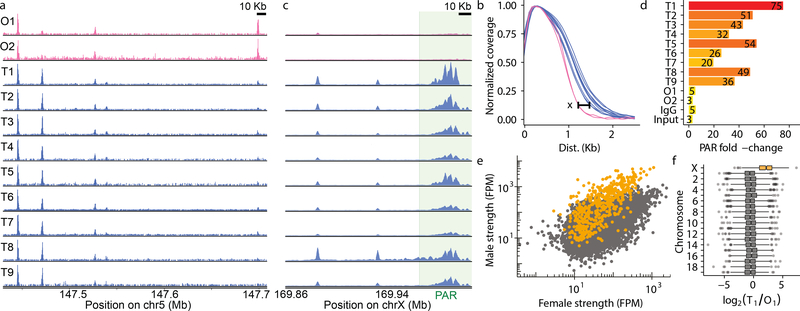
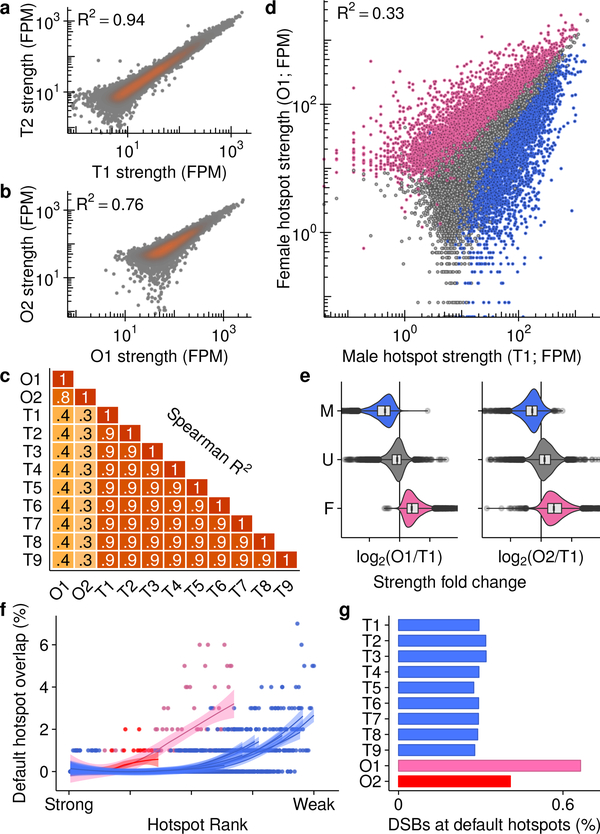
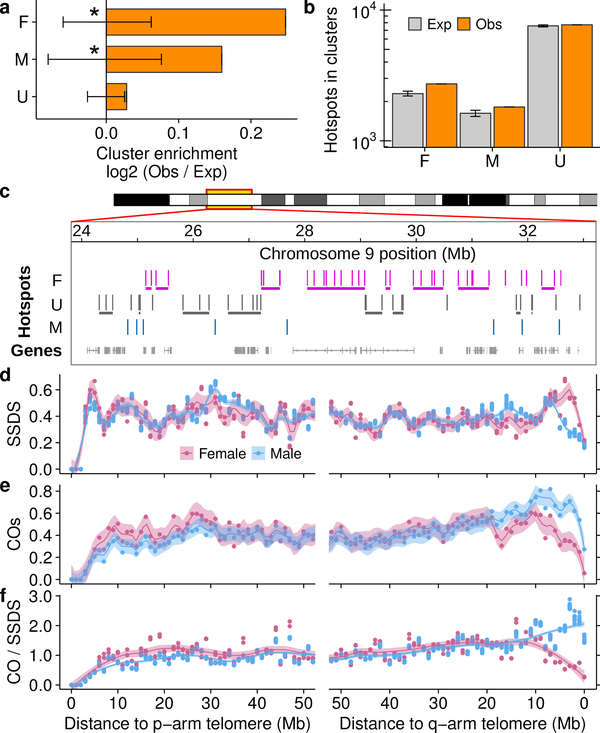
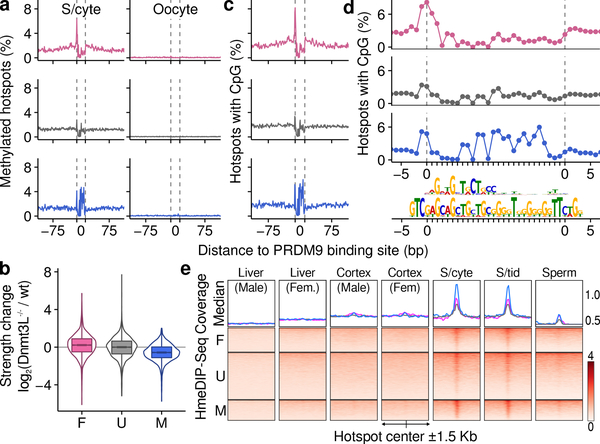
Meiotic recombination differs between males and females; however, when and how these differences are established is unknown. Here we identify extensive sex differences at the initiation of recombination by mapping hotspots of meiotic DNA double-strand breaks in male and female mice. Contrary to past findings in humans, few hotspots are used uniquely in either sex. Instead, grossly different recombination landscapes result from up to fifteen-fold differences in hotspot usage between males and females. Indeed, most recombination occurs at sex-biased hotspots. Sex-biased hotspots seem to be partly determined by chromosome structure, and DNA methylation, which is absent in females at the onset of meiosis, has a substantial role. Sex differences are also evident later in meiosis as the rate at which meiotic breaks are repaired as crossovers differs between males and females in distal regions. The suppression of distal crossovers may help to minimize age-related aneuploidy that arises owing to cohesion loss during dictyate arrest in females.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
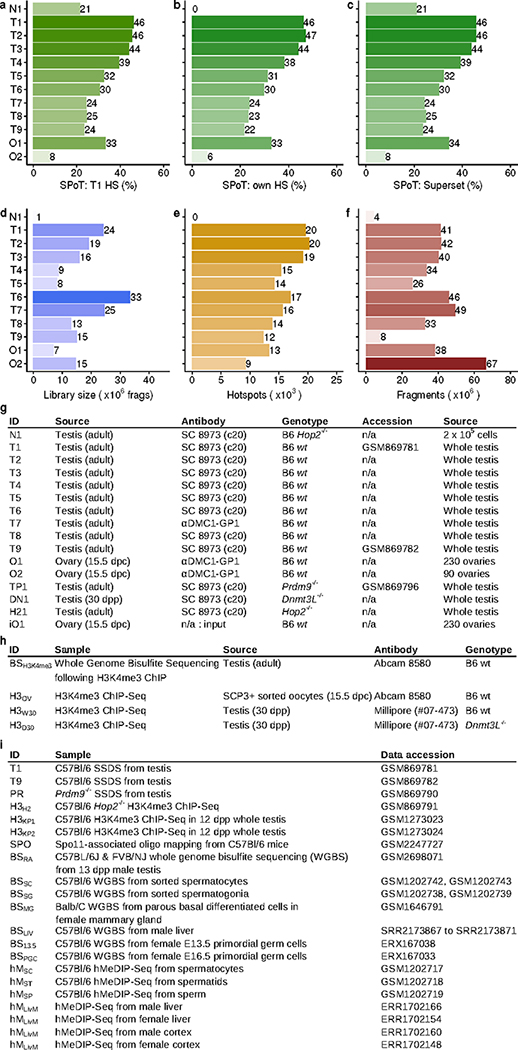
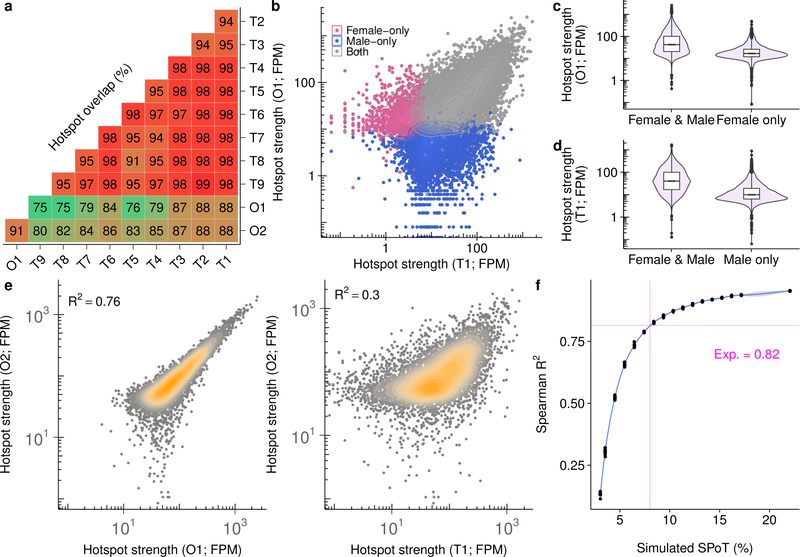

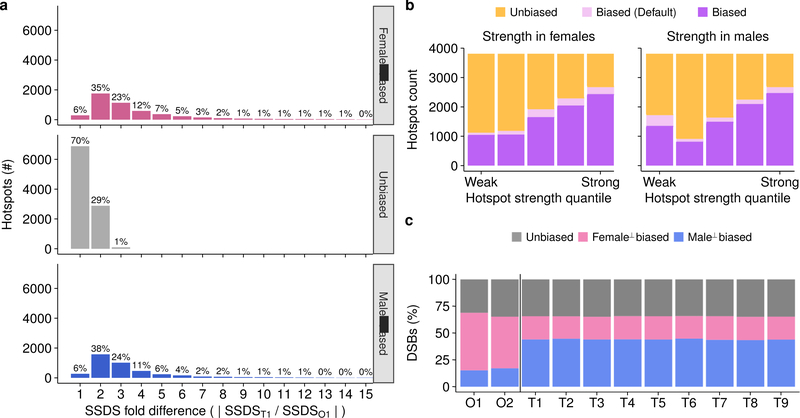
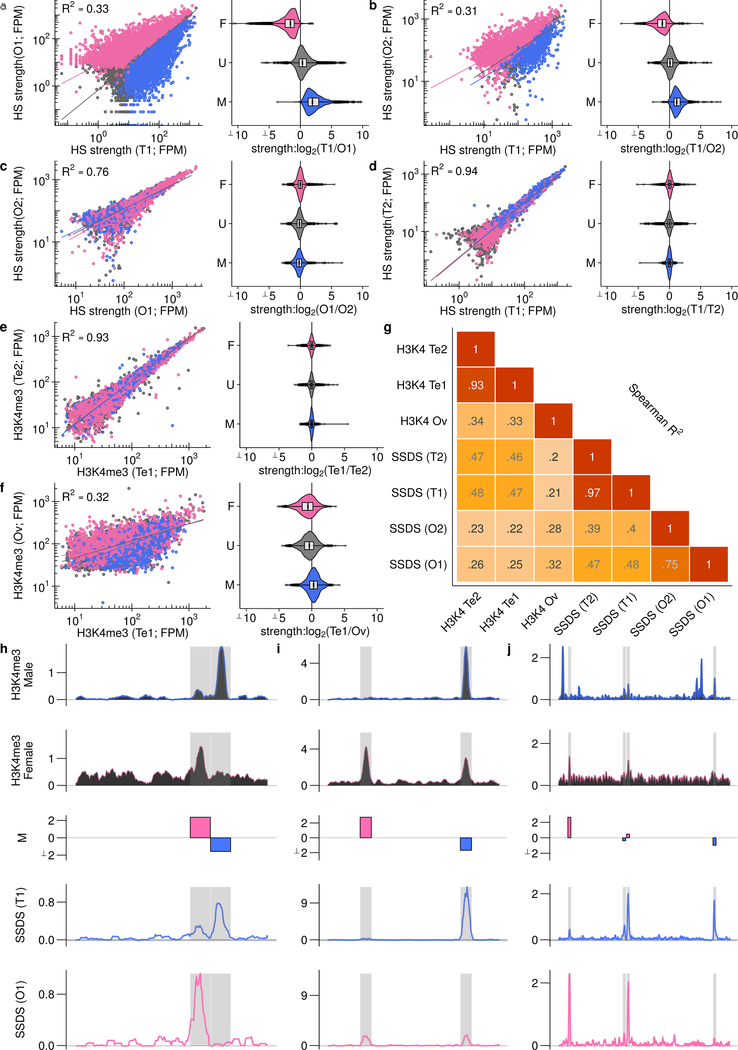
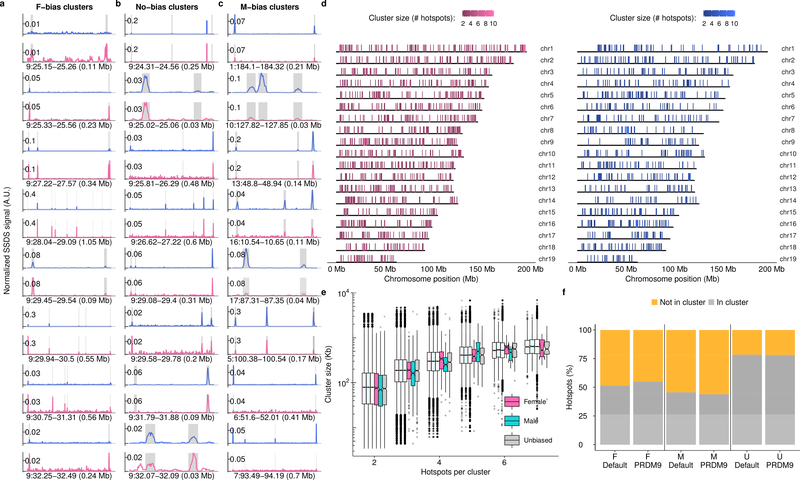
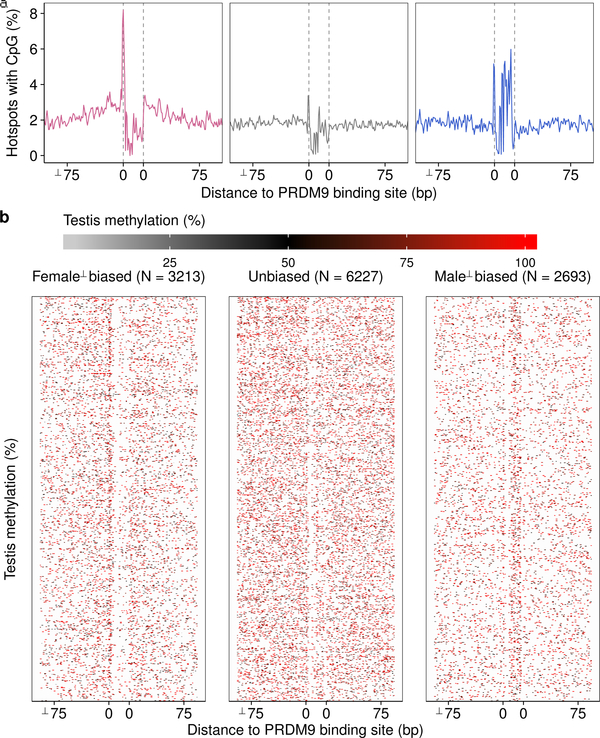
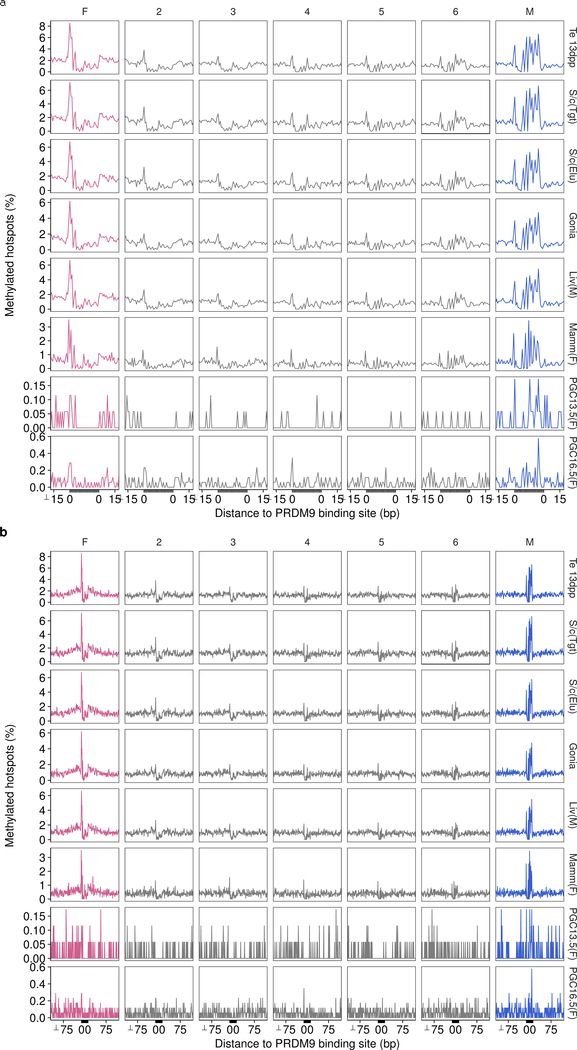
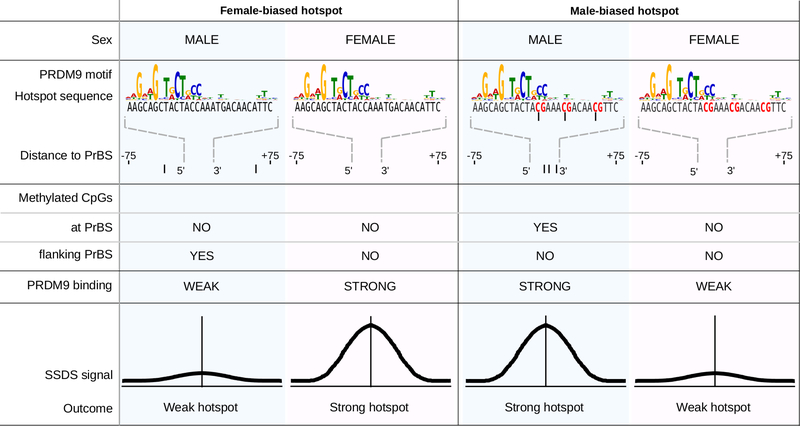
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
