

Impaired Glycine Receptor Trafficking in Neurological Diseases
- PMID: 30186111
- PMCID: PMC6110938
- DOI: 10.3389/fnmol.2018.00291
Impaired Glycine Receptor Trafficking in Neurological Diseases
Abstract
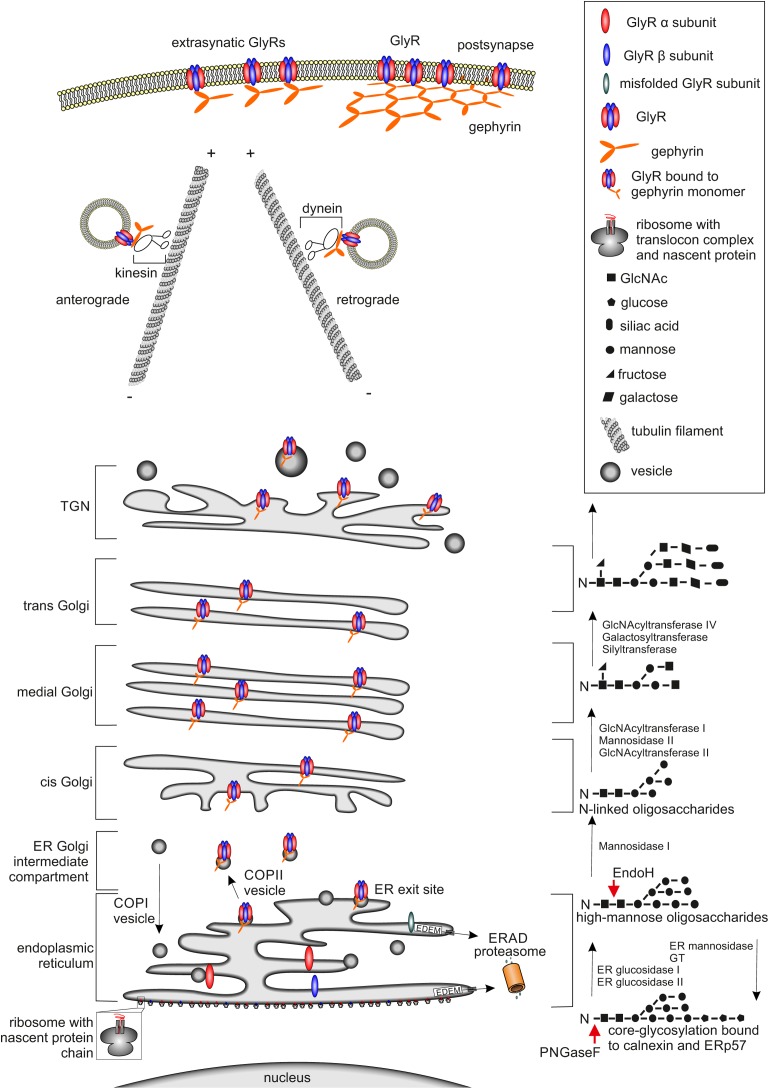
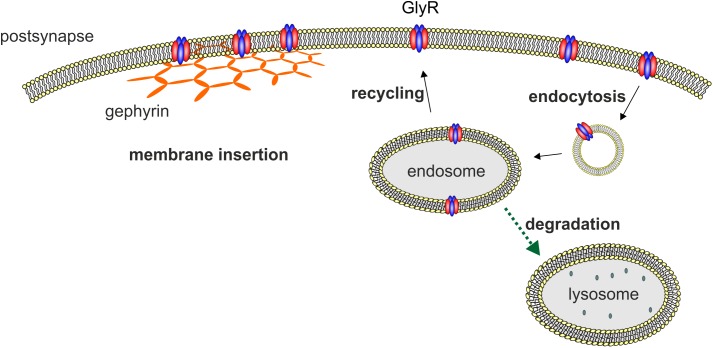
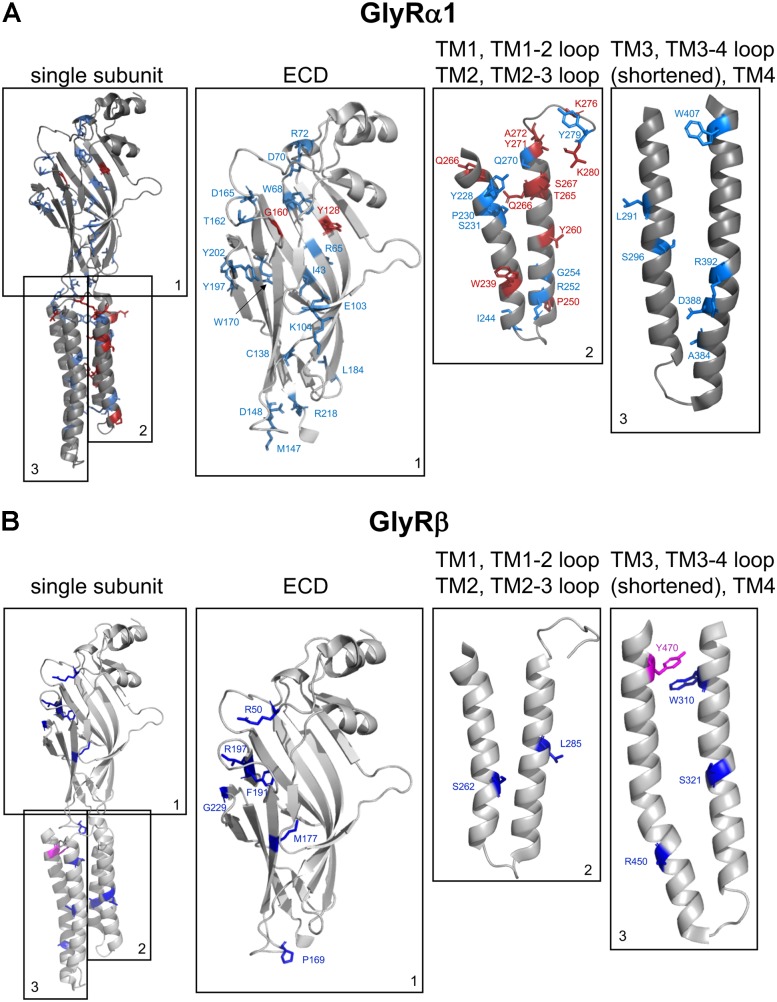
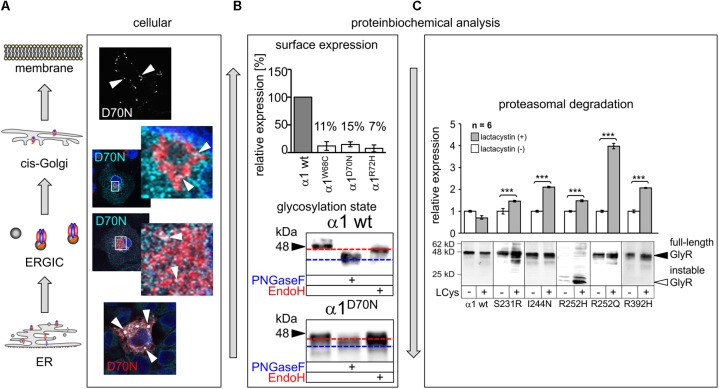
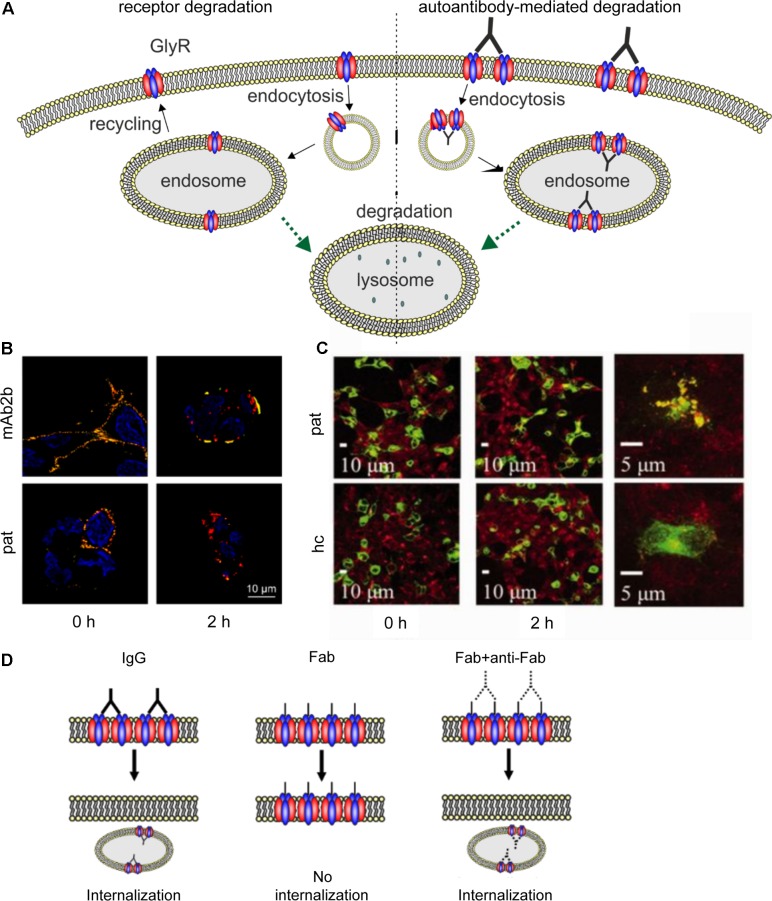
Ionotropic glycine receptors (GlyRs) enable fast synaptic neurotransmission in the adult spinal cord and brainstem. The inhibitory GlyR is a transmembrane glycine-gated chloride channel. The immature GlyR protein undergoes various processing steps, e.g., folding, assembly, and maturation while traveling from the endoplasmic reticulum to and through the Golgi apparatus, where post-translational modifications, e.g., glycosylation occur. The mature receptors are forward transported via microtubules to the cellular surface and inserted into neuronal membranes followed by synaptic clustering. The normal life cycle of a receptor protein includes further processes like internalization, recycling, and degradation. Defects in GlyR life cycle, e.g., impaired protein maturation and degradation have been demonstrated to underlie pathological mechanisms of various neurological diseases. The neurological disorder startle disease is caused by glycinergic dysfunction mainly due to missense mutations in genes encoding GlyR subunits (GLRA1 and GLRB). In vitro studies have shown that most recessive forms of startle disease are associated with impaired receptor biogenesis. Another neurological disease with a phenotype similar to startle disease is a special form of stiff-person syndrome (SPS), which is most probably due to the development of GlyR autoantibodies. Binding of GlyR autoantibodies leads to enhanced receptor internalization. Here we focus on the normal life cycle of GlyRs concentrating on assembly and maturation, receptor trafficking, post-synaptic integration and clustering, and GlyR internalization/recycling/degradation. Furthermore, this review highlights findings on impairment of these processes under disease conditions such as disturbed neuronal ER-Golgi trafficking as the major pathomechanism for recessive forms of human startle disease. In SPS, enhanced receptor internalization upon autoantibody binding to the GlyR has been shown to underlie the human pathology. In addition, we discuss how the existing mouse models of startle disease increased our current knowledge of GlyR trafficking routes and function. This review further illuminates receptor trafficking of GlyR variants originally identified in startle disease patients and explains changes in the life cycle of GlyRs in patients with SPS with respect to structural and functional consequences at the receptor level.
Keywords: autoimmune antibodies; glycine receptor; protein maturation; startle disease; trafficking pathways.
Figures
References
-
- Atak S., Langlhofer G., Schaefer N., Kessler D., Meiselbach H., Delto C., et al. (2015). Disturbances of ligand potency and enhanced degradation of the human glycine receptor at affected positions G160 and T162 originally identified in patients suffering from hyperekplexia. Front. Mol. Neurosci. 8:79. 10.3389/fnmol.2015.00079 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources