

Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview
- PMID: 30186166
- PMCID: PMC6113859
- DOI: 10.3389/fphar.2018.00923
Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview
Abstract
Computational techniques have been applied in the drug discovery pipeline since the 1980s. Given the low computational resources of the time, the first molecular modeling strategies relied on a rigid view of the ligand-target binding process. During the years, the evolution of hardware technologies has gradually allowed simulating the dynamic nature of the binding event. In this work, we present an overview of the evolution of structure-based drug discovery techniques in the study of ligand-target recognition phenomenon, going from the static molecular docking toward enhanced molecular dynamics strategies.
Keywords: enhanced sampling; ligand-protein binding; molecular docking; molecular dynamics; molecular recognition; protein flexibility.
Figures
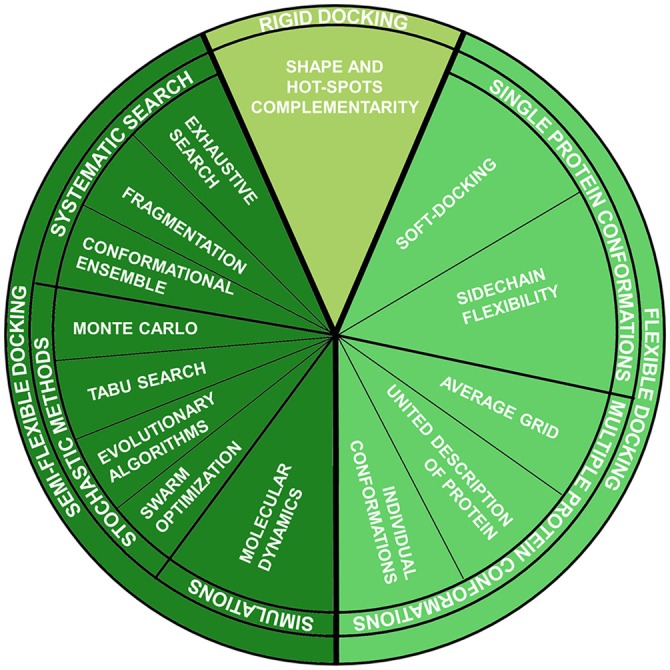
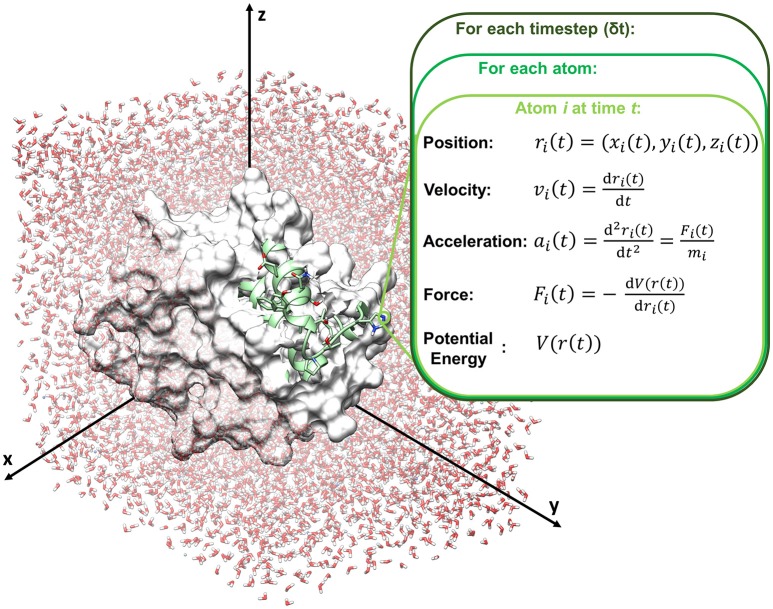
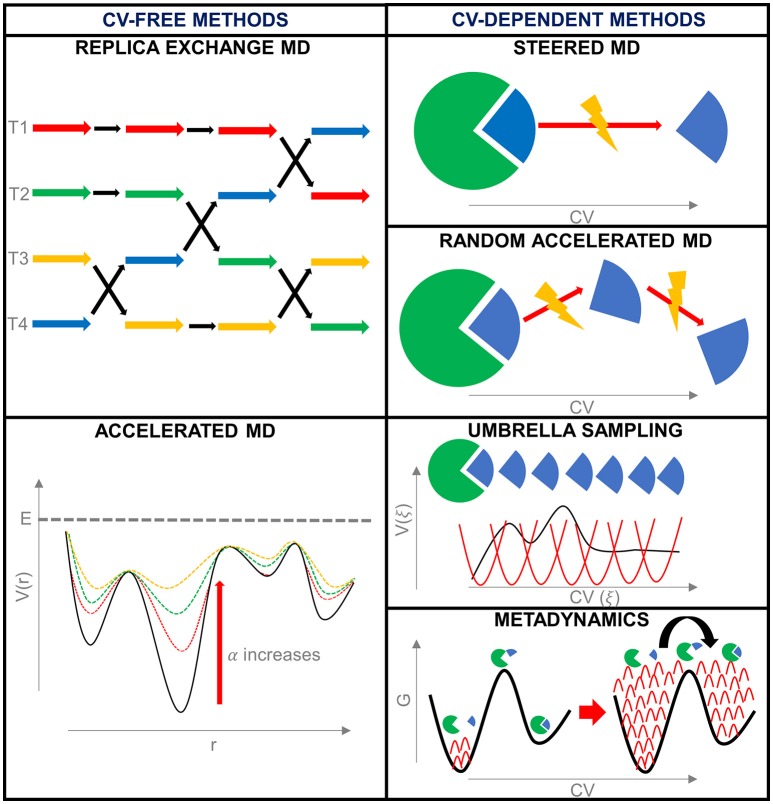
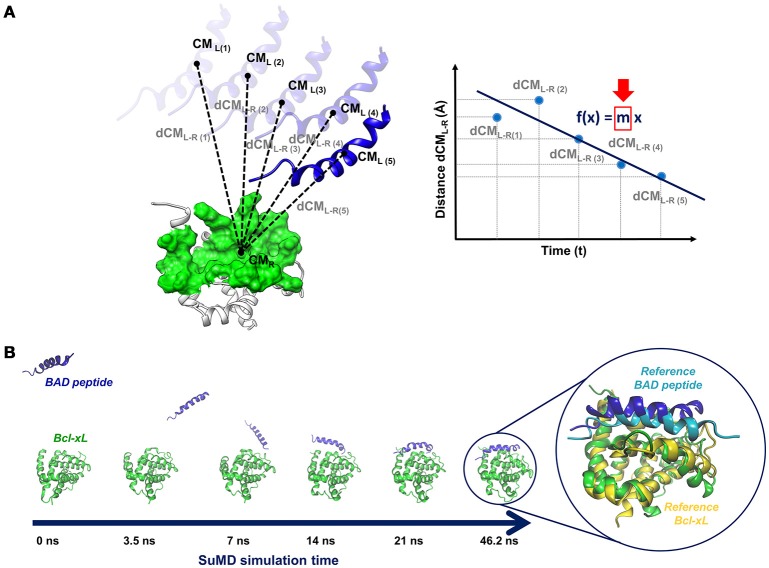
References
-
- Abagyan R., Totrov M., Kuznetsov D. (1994). ICM? A new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506. 10.1002/jcc.540150503 - DOI
-
- Alder B. J., Wainwright T. E. (1957). Phase transition for a hard sphere system. J. Chem. Phys. 27, 1208–1209. 10.1063/1.1743957 - DOI
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources