

Nuclear Speckle RNA Binding Proteins Remodel Alternative Splicing and the Non-coding Arabidopsis Transcriptome to Regulate a Cross-Talk Between Auxin and Immune Responses
- PMID: 30186296
- PMCID: PMC6111844
- DOI: 10.3389/fpls.2018.01209
Nuclear Speckle RNA Binding Proteins Remodel Alternative Splicing and the Non-coding Arabidopsis Transcriptome to Regulate a Cross-Talk Between Auxin and Immune Responses
Abstract
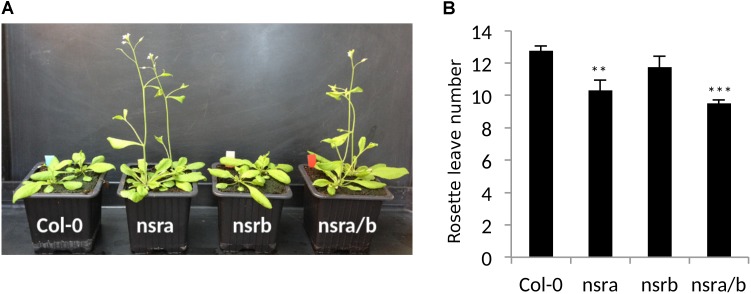
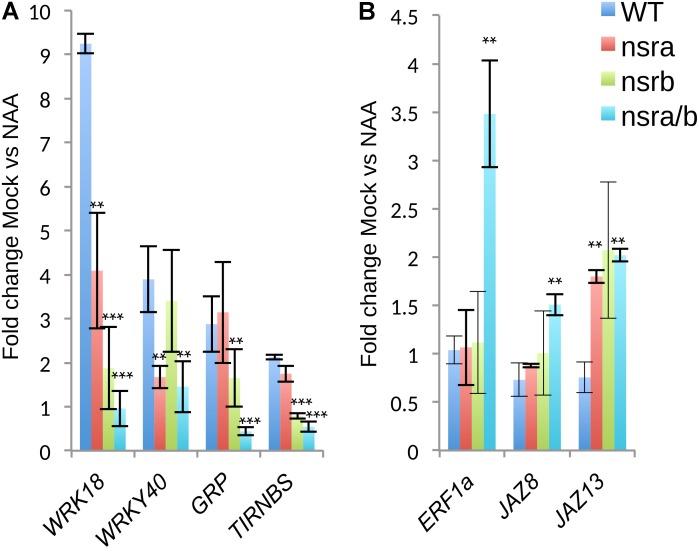
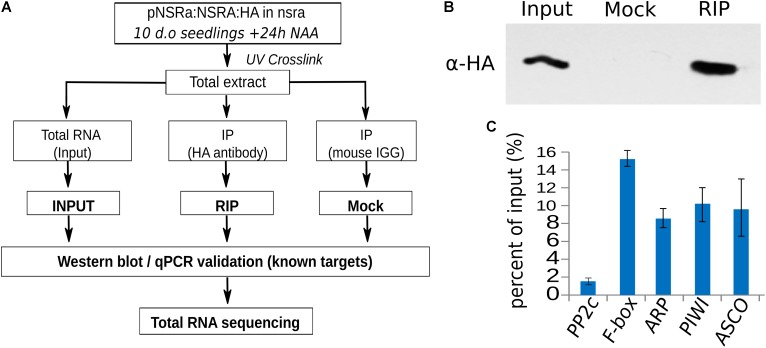
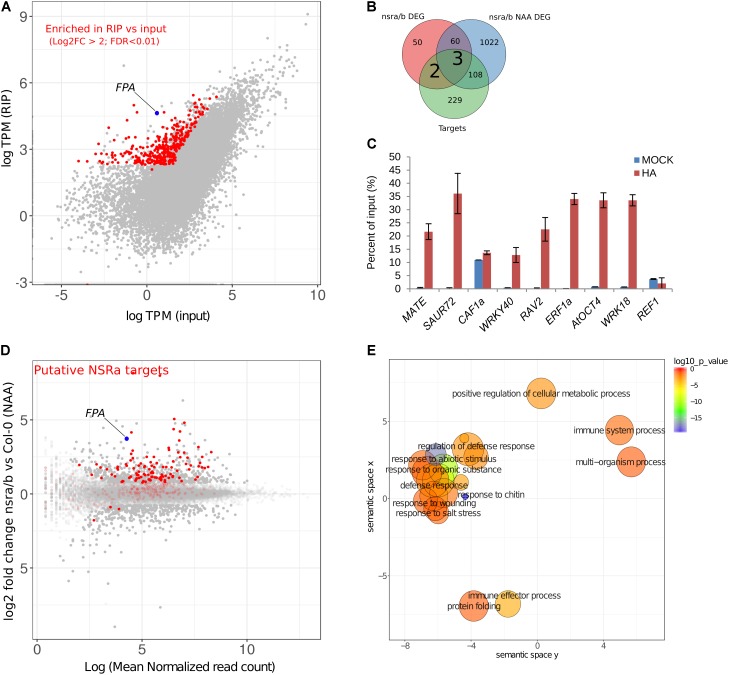
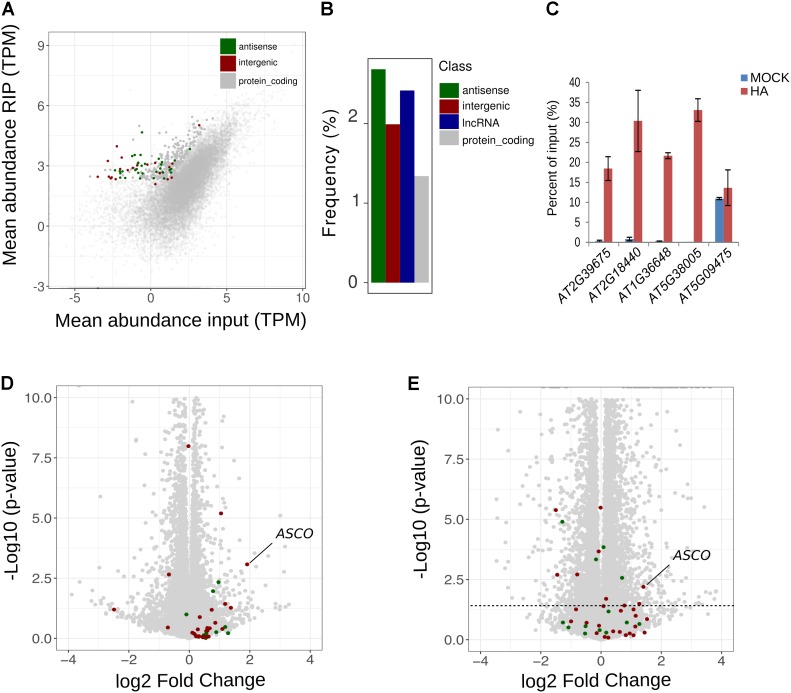
Nuclear speckle RNA binding proteins (NSRs) act as regulators of alternative splicing (AS) and auxin-regulated developmental processes such as lateral root formation in Arabidopsis thaliana. These proteins were shown to interact with specific alternatively spliced mRNA targets and at least with one structured lncRNA, named Alternative Splicing Competitor RNA. Here, we used genome-wide analysis of RNAseq to monitor the NSR global role on multiple tiers of gene expression, including RNA processing and AS. NSRs affect AS of 100s of genes as well as the abundance of lncRNAs particularly in response to auxin. Among them, the FPA floral regulator displayed alternative polyadenylation and differential expression of antisense COOLAIR lncRNAs in nsra/b mutants. This may explains the early flowering phenotype observed in nsra and nsra/b mutants. GO enrichment analysis of affected lines revealed a novel link of NSRs with the immune response pathway. A RIP-seq approach on an NSRa fusion protein in mutant background identified that lncRNAs are privileged direct targets of NSRs in addition to specific AS mRNAs. The interplay of lncRNAs and AS mRNAs in NSR-containing complexes may control the crosstalk between auxin and the immune response pathway.
Keywords: RNA binding proteins; RNP complexes; alternative splicing; auxin; immune response.
Figures





References
-
- Andrews S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous