

Phylogenomic analysis unravels evolution of yellow fever virus within hosts
- PMID: 30188905
- PMCID: PMC6143276
- DOI: 10.1371/journal.pntd.0006738
Phylogenomic analysis unravels evolution of yellow fever virus within hosts
Abstract
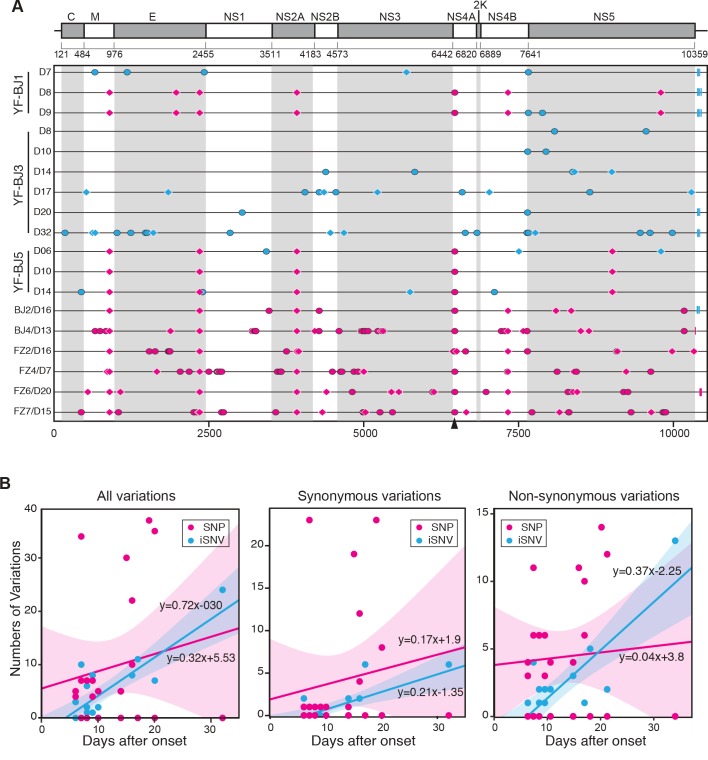
The yellow fever virus (YFV) recently reemerged in the large outbreaks in Africa and Brazil, and the first imported patients into Asia have recalled the concerns of YFV evolution. Here we show phylogenomics of YFV with serial clinical samples of the 2016 YFV infections. Phylogenetics exhibited that the 2016 strains were close to Angola 1971 strains and only three amino acid changes presented new to other lineages. Deep sequencing of viral genomes discovered 101 intrahost single nucleotide variations (iSNVs) and 234 single nucleotide polymorphisms (SNPs). Analysis of iSNV distribution and mutated allele frequency revealed that the coding regions were under purifying selection. Comparison of the evolutionary rates estimated by iSNV and SNP showed that the intrahost rate was ~2.25 times higher than the epidemic rate, and both rates were higher than the long-term YFV substitution rate, as expected. In addition, the result also hinted that short viremia duration of YFV might further hinder the evolution of YFV.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
-
- Mutebi J-P, Rijnbrand RC, Wang H, et al. Genetic relationships and evolution of genotypes of yellow fever virus and other members of the yellow fever virus group within the Flavivirus genus based on the 3′ noncoding region. Journal of virology 2004; 78:9652–65. 10.1128/JVI.78.18.9652-9665.2004 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
