

Wilson disease
- PMID: 30190489
- PMCID: PMC6416051
- DOI: 10.1038/s41572-018-0018-3
Wilson disease
Abstract
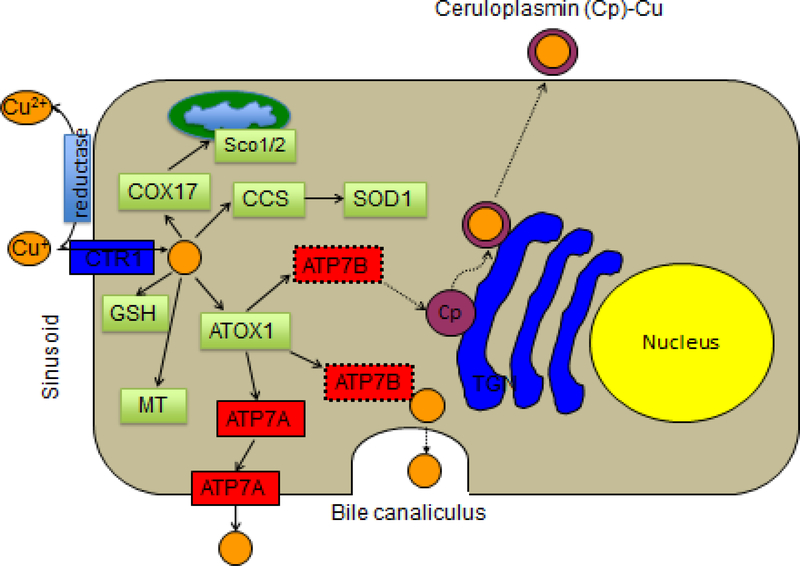
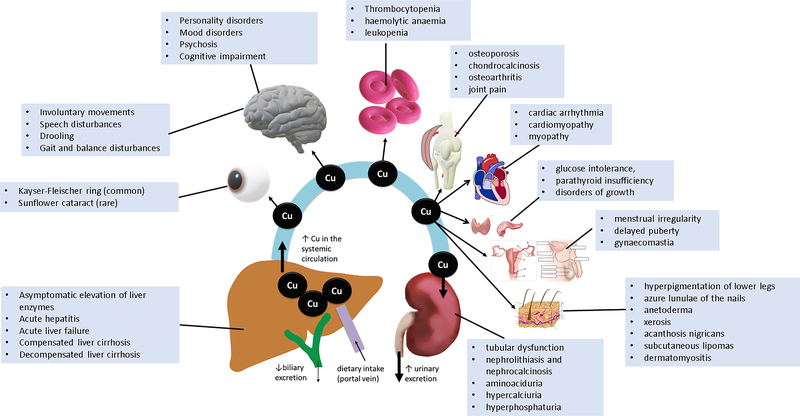
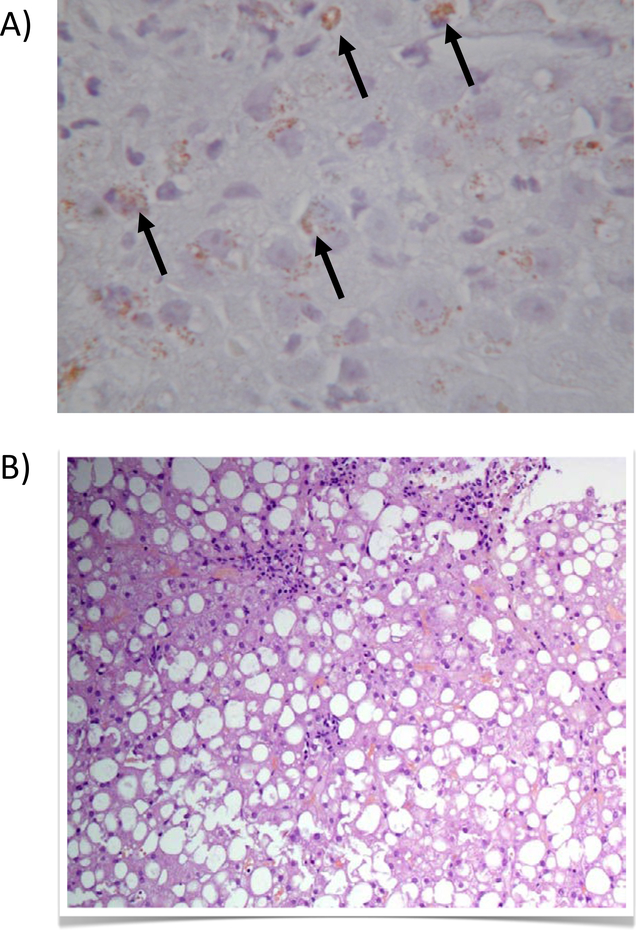
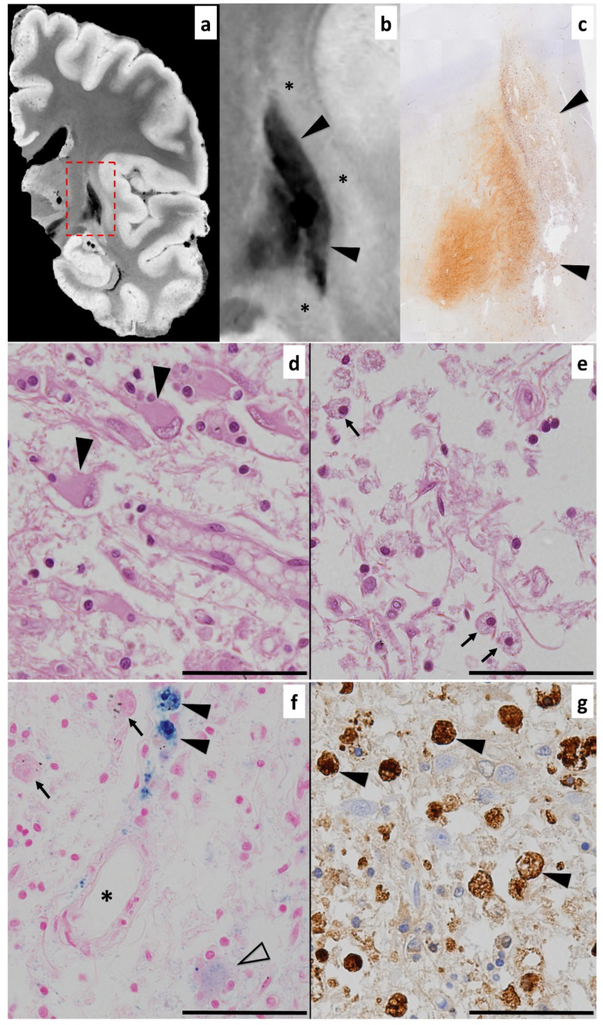
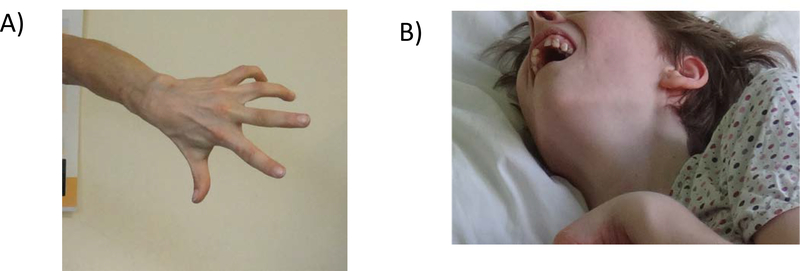
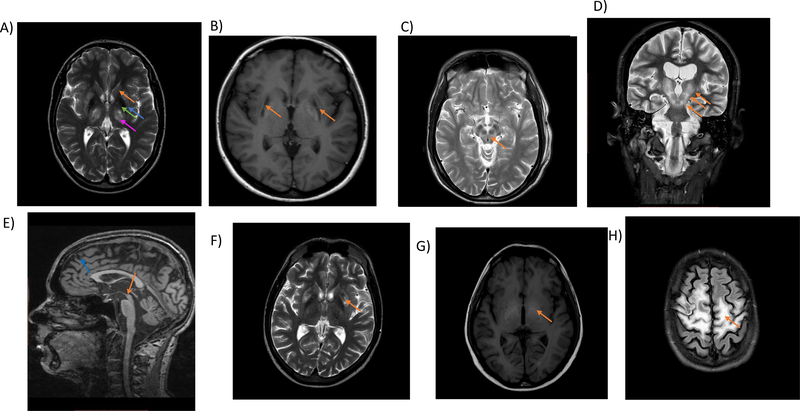
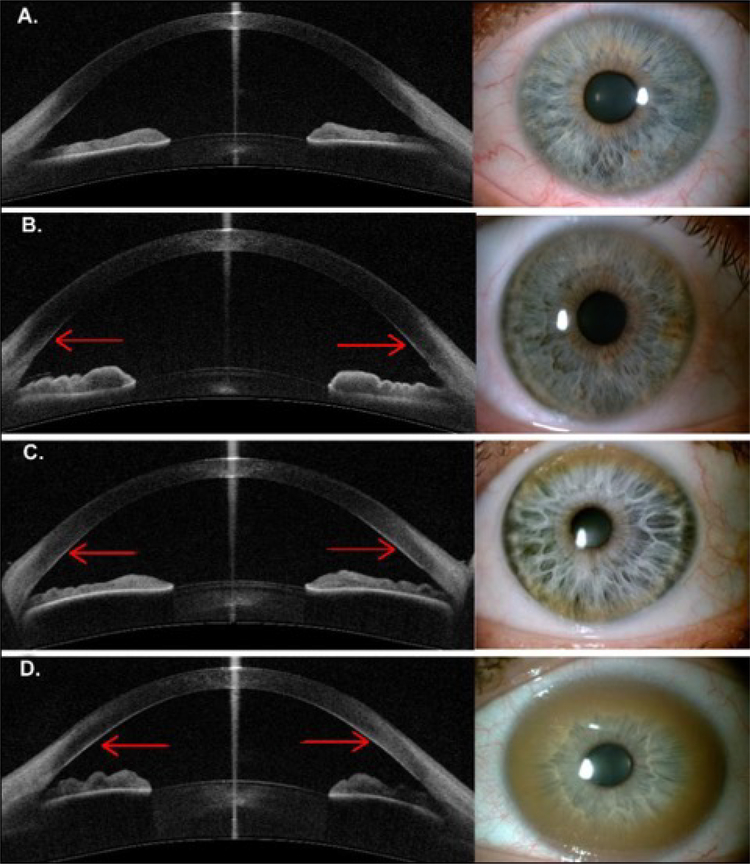
Wilson disease (WD) is a potentially treatable, inherited disorder of copper metabolism that is characterized by the pathological accumulation of copper. WD is caused by mutations in ATP7B, which encodes a transmembrane copper-transporting ATPase, leading to impaired copper homeostasis and copper overload in the liver, brain and other organs. The clinical course of WD can vary in the type and severity of symptoms, but progressive liver disease is a common feature. Patients can also present with neurological disorders and psychiatric symptoms. WD is diagnosed using diagnostic algorithms that incorporate clinical symptoms and signs, measures of copper metabolism and DNA analysis of ATP7B. Available treatments include chelation therapy and zinc salts, which reverse copper overload by different mechanisms. Additionally, liver transplantation is indicated in selected cases. New agents, such as tetrathiomolybdate salts, are currently being investigated in clinical trials, and genetic therapies are being tested in animal models. With early diagnosis and treatment, the prognosis is good; however, an important issue is diagnosing patients before the onset of serious symptoms. Advances in screening for WD may therefore bring earlier diagnosis and improvements for patients with WD.
Conflict of interest statement
Competing interests
A.C. has served on advisory boards for Wilson Therapeutics, Vivet Therapeutics, GMPO and received speaker fees from EVER, Boehringer Ingelheim and Nutricia; P.F. has served on advisory boards for Wilson Therapeutics, Vivet Therapeutics and Univar and received speaker fees from Univar; V.M. has served as a consultant for Kadmon; K.H.W. is on speakers bureau of Abbvie, Alexion, Bayer, BMS, Chiesi, GMPO, Norgine, Novartis, Univar, Wilson Therapeutics, Vivet Therapeutics and received grants (to the institution) from Alexion, Bayer, BMS, Eisai, GMPO, Novartis, Univar and Wilson Therapeutics; M.L.S. has served on advisory boards for Wilson Therapeutics, Vivet Therapeutics, GMPO and Kadmon, is a speaker for Gilead and is on the Medical Advisory Committee of the Wilson disease association; T.L., P.D., S.L. and J.K.R. declare no competing interests.
Figures

References
-
- Ferenci P Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet 120, 151–159 (2006). - PubMed
-
- Dzieżyc K, Karliński M, Litwin T & Członkowska A Compliant treatment with anti-copper agents prevents clinically overt Wilson’s disease in pre-symptomatic patients. Eur. J. Neurol 21, 332–337 (2013). - PubMed
-
- EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol 56, 671–685 (2012). - PubMed
-
- Roberts EA & Schilsky ML Diagnosis and treatment of Wilson disease: An update. Hepatology 47, 2089–2111 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
