

Immuno-Oncology: Emerging Targets and Combination Therapies
- PMID: 30191140
- PMCID: PMC6115503
- DOI: 10.3389/fonc.2018.00315
Immuno-Oncology: Emerging Targets and Combination Therapies
Abstract
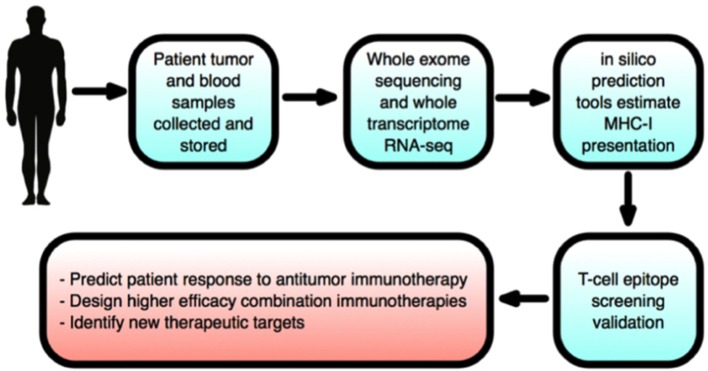
Host immunity recognizes and eliminates most early tumor cells, yet immunological checkpoints, exemplified by CTLA-4, PD-1, and PD-L1, pose a significant obstacle to effective antitumor immune responses. T-lymphocyte co-inhibitory pathways influence intensity, inflammation and duration of antitumor immunity. However, tumors and their immunosuppressive microenvironments exploit them to evade immune destruction. Recent PD-1 checkpoint inhibitors yielded unprecedented efficacies and durable responses across advanced-stage melanoma, showcasing potential to replace conventional radiotherapy regimens. Neverthless, many clinical problems remain in terms of efficacy, patient-to-patient variability, and undesirable outcomes and side effects. In this review, we evaluate recent advances in the immuno-oncology field and discuss ways forward. First, we give an overview of current immunotherapy modalities, involving mainy single agents, including inhibitor monoclonal antibodies (mAbs) targeting T-cell checkpoints of PD-1 and CTLA-4. However, neoantigen recognition alone cannot eliminate tumors effectively in vivo given their inherent complex micro-environment, heterogeneous nature and stemness. Then, based mainly upon CTLA-4 and PD-1 checkpoint inhibitors as a "backbone," we cover a range of emerging ("second-generation") therapies incorporating other immunotherapies or non-immune based strategies in synergistic combination. These include targeted therapies such as tyrosine kinase inhibitors, co-stimulatory mAbs, bifunctional agents, epigenetic modulators (such as inhibitors of histone deacetylases or DNA methyltransferase), vaccines, adoptive-T-cell therapy, nanoparticles, oncolytic viruses, and even synthetic "gene circuits." A number of novel immunotherapy co-targets in pre-clinical development are also introduced. The latter include metabolic components, exosomes and ion channels. We discuss in some detail of the personalization of immunotherapy essential for ultimate maximization of clinical outcomes. Finally, we outline possible future technical and conceptual developments including realistic in vitro and in vivo models and inputs from physics, engineering, and artificial intelligence. We conclude that the breadth and quality of immunotherapeutic approaches and the types of cancers that can be treated will increase significantly in the foreseeable future.
Keywords: biomarkers; checkpoint blockade; combination immunotherapy; personalized therapy; tumor microenvironment.
Figures









References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials