

The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment
- PMID: 30193109
- PMCID: PMC6136429
- DOI: 10.1016/j.cell.2018.08.027
The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment
Abstract
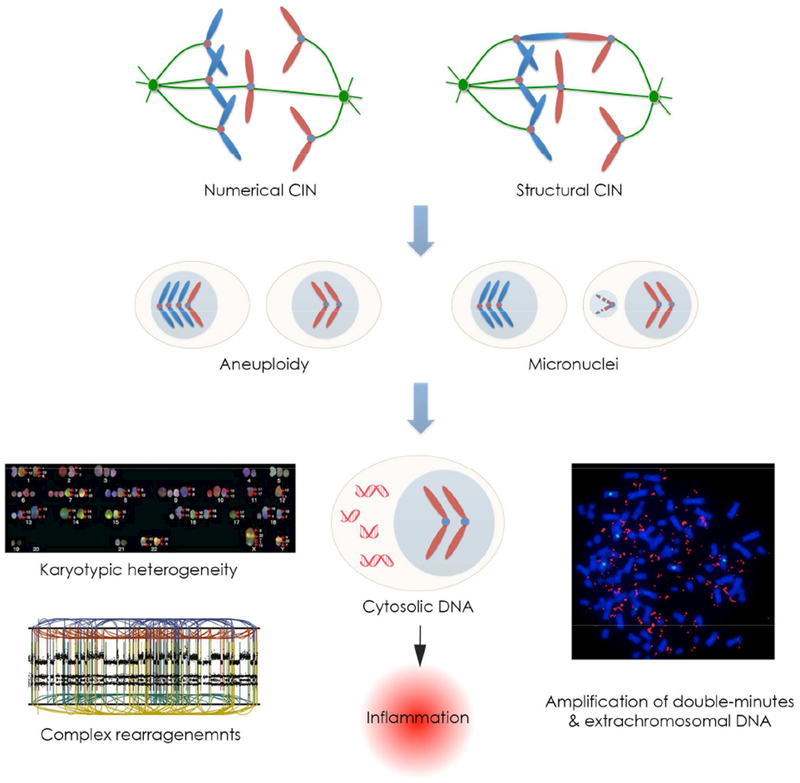
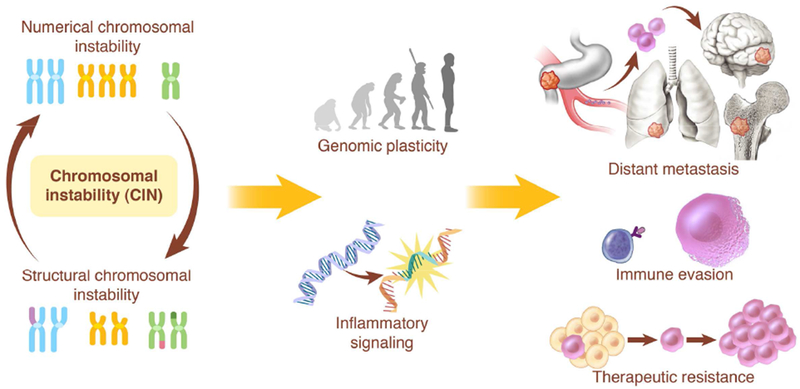
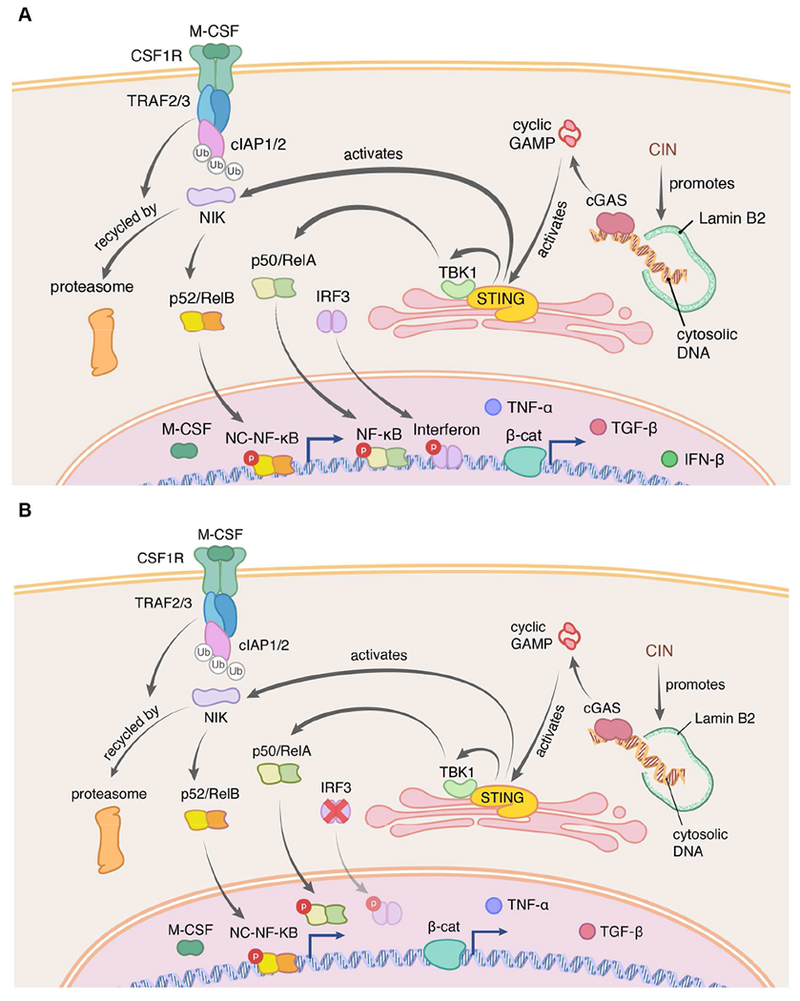
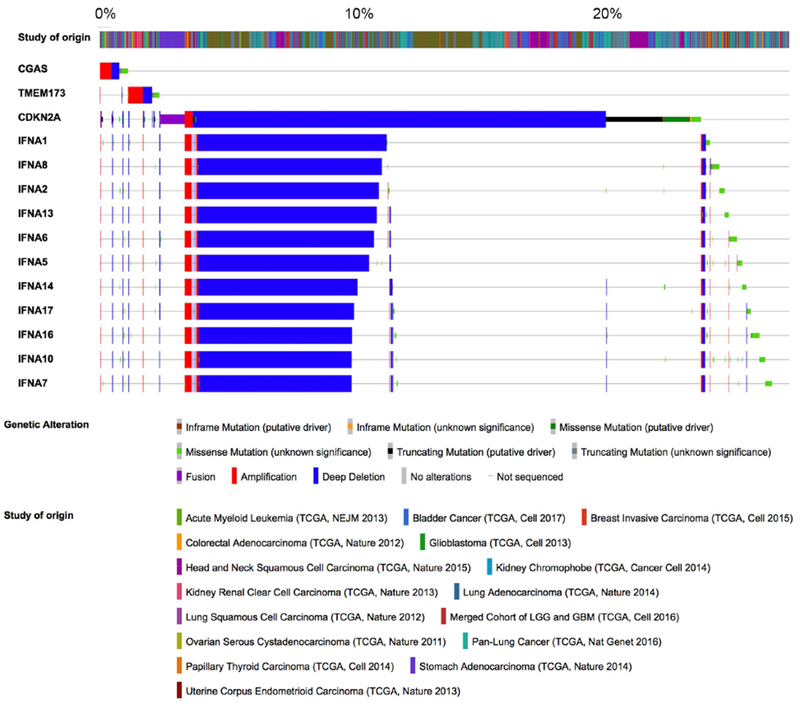
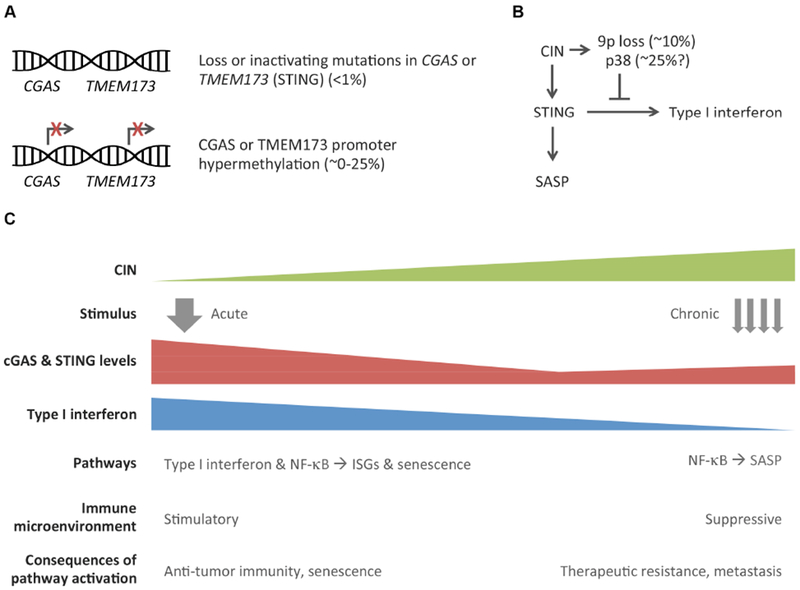
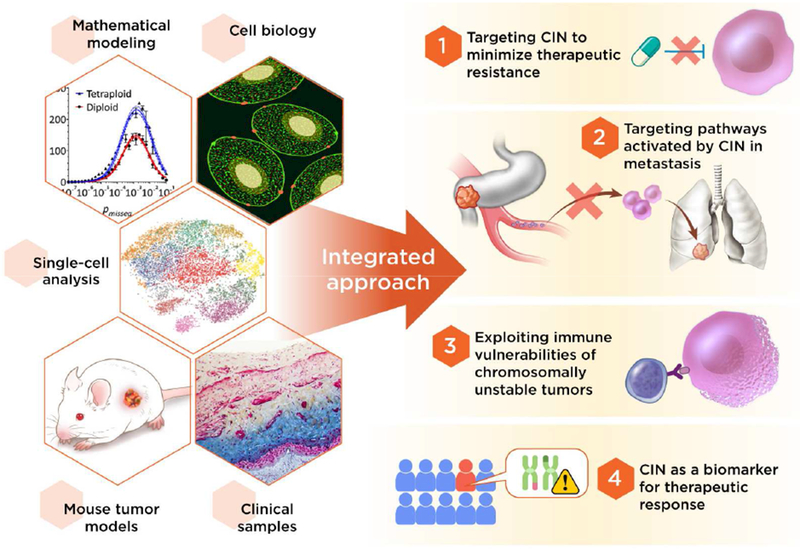
Chromosomal instability (CIN) is a hallmark of human cancer, and it is associated with poor prognosis, metastasis, and therapeutic resistance. CIN results from errors in chromosome segregation during mitosis, leading to structural and numerical chromosomal abnormalities. In addition to generating genomic heterogeneity that acts as a substrate for natural selection, CIN promotes inflammatory signaling by introducing double-stranded DNA into the cytosol, engaging the cGAS-STING anti-viral pathway. These multipronged effects distinguish CIN as a central driver of tumor evolution and as a genomic source for the crosstalk between the tumor and its microenvironment, in the course of immune editing and evasion.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
S.F.B. declares no competing interests. L.C.C. owns equity in, receives compensation from, and serves on the board of directors and scientific advisory board of Agios Pharmaceuticals. He is also a founder of and receives laboratory support from Petra Pharmaceuticals.
Figures
References
-
- Ame JC, Cimini D, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, and Schreiber V (2008). Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim. Biophys. Acta 1786, 32–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
