

Genomic Prediction Using Multi-trait Weighted GBLUP Accounting for Heterogeneous Variances and Covariances Across the Genome
- PMID: 30194089
- PMCID: PMC6222589
- DOI: 10.1534/g3.118.200673
Genomic Prediction Using Multi-trait Weighted GBLUP Accounting for Heterogeneous Variances and Covariances Across the Genome
Abstract
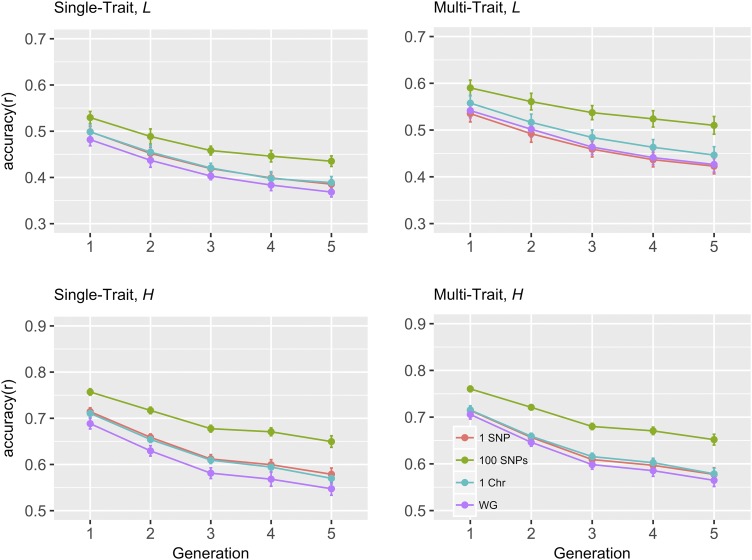
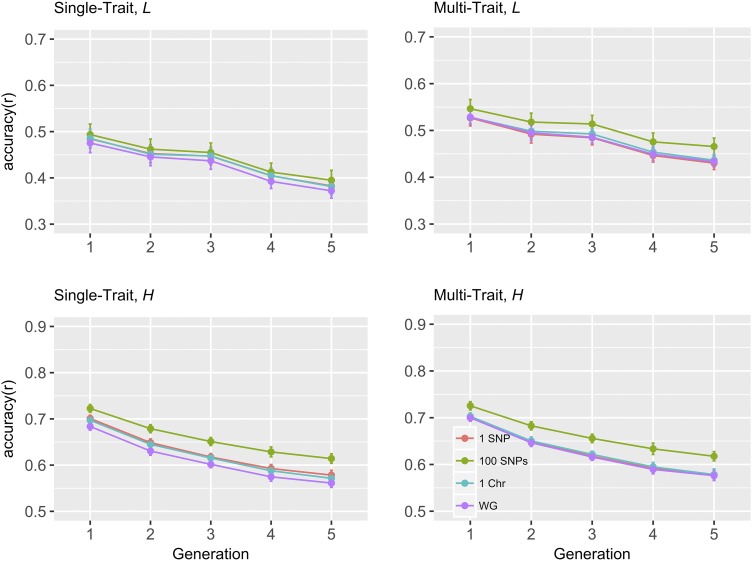
Implicit assumption of common (co)variance for all loci in multi-trait Genomic Best Linear Unbiased Prediction (GBLUP) results in a genomic relationship matrix (G) that is common to all traits. When this assumption is violated, Bayesian whole genome regression methods may be superior to GBLUP by accounting for unequal (co)variance for all loci or genome regions. This study aimed to develop a strategy to improve the accuracy of GBLUP for multi-trait genomic prediction, using (co)variance estimates of SNP effects from Bayesian whole genome regression methods. Five generations (G1-G5, test populations) of genotype data were available by simulations based on data of 2,200 Danish Holstein cows (G0, reference population). Two correlated traits with heritabilities of 0.1 or 0.4, and a genetic correlation of 0.45 were generated. First, SNP effects and breeding values were estimated using BayesAS method, assuming (co)variance was the same for SNPs within a genome region, and different between regions. Region size was set as one SNP, 100 SNPs, a whole chromosome or whole genome. Second, posterior (co)variances of SNP effects were used to weight SNPs in construction of G matrices. In general, region size of 100 SNPs led to highest prediction accuracies using BayesAS, and wGBLUP outperformed GBLUP at this region size. Our results suggest that when genetic architectures of traits favor Bayesian methods, the accuracy of multi-trait GBLUP can be as high as the Bayesian method if SNPs are weighted by the Bayesian posterior (co)variances.
Keywords: GenPred; Genetic architecture; Genomic prediction; Genomic relationship matrix; Region size; SNP weight; Shared Data Resources.
Copyright © 2018 Karaman et al.
Figures
Similar articles
-
Modeling heterogeneous (co)variances from adjacent-SNP groups improves genomic prediction for milk protein composition traits.Genet Sel Evol. 2017 Dec 5;49(1):89. doi: 10.1186/s12711-017-0364-8. Genet Sel Evol. 2017. PMID: 29207947 Free PMC article.
-
Multi-trait single-step genomic prediction accounting for heterogeneous (co)variances over the genome.Heredity (Edinb). 2020 Feb;124(2):274-287. doi: 10.1038/s41437-019-0273-4. Epub 2019 Oct 22. Heredity (Edinb). 2020. PMID: 31641237 Free PMC article.
-
The patterns of genomic variances and covariances across genome for milk production traits between Chinese and Nordic Holstein populations.BMC Genet. 2017 Mar 15;18(1):26. doi: 10.1186/s12863-017-0491-9. BMC Genet. 2017. PMID: 28298201 Free PMC article.
-
Application of Bayesian genomic prediction methods to genome-wide association analyses.Genet Sel Evol. 2022 May 13;54(1):31. doi: 10.1186/s12711-022-00724-8. Genet Sel Evol. 2022. PMID: 35562659 Free PMC article. Review.
-
Improving Genomic Prediction Using High-Dimensional Secondary Phenotypes.Front Genet. 2021 May 24;12:667358. doi: 10.3389/fgene.2021.667358. eCollection 2021. Front Genet. 2021. PMID: 34108993 Free PMC article. Review.
Cited by
-
(Quasi) multitask support vector regression with heuristic hyperparameter optimization for whole-genome prediction of complex traits: a case study with carcass traits in broilers.G3 (Bethesda). 2023 Aug 9;13(8):jkad109. doi: 10.1093/g3journal/jkad109. G3 (Bethesda). 2023. PMID: 37216670 Free PMC article.
-
Weighted single-step genomic best linear unbiased prediction integrating variants selected from sequencing data by association and bioinformatics analyses.Genet Sel Evol. 2020 Aug 14;52(1):48. doi: 10.1186/s12711-020-00568-0. Genet Sel Evol. 2020. PMID: 32799816 Free PMC article.
-
Genetic Strategies for Enhancing Rooster Fertility in Tropical and Humid Climates: Challenges and Opportunities.Animals (Basel). 2025 Apr 10;15(8):1096. doi: 10.3390/ani15081096. Animals (Basel). 2025. PMID: 40281930 Free PMC article. Review.
-
Genomic prediction using a reference population of multiple pure breeds and admixed individuals.Genet Sel Evol. 2021 May 31;53(1):46. doi: 10.1186/s12711-021-00637-y. Genet Sel Evol. 2021. PMID: 34058971 Free PMC article.
-
Quercus species divergence is driven by natural selection on evolutionarily less integrated traits.Heredity (Edinb). 2021 Feb;126(2):366-382. doi: 10.1038/s41437-020-00378-6. Epub 2020 Oct 27. Heredity (Edinb). 2021. PMID: 33110229 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources