

Necroptosis mediates myofibre death in dystrophin-deficient mice
- PMID: 30194302
- PMCID: PMC6128848
- DOI: 10.1038/s41467-018-06057-9
Necroptosis mediates myofibre death in dystrophin-deficient mice
Erratum in
-
Publisher Correction: Necroptosis mediates myofibre death in dystrophin-deficient mice.Nat Commun. 2018 Oct 2;9(1):4107. doi: 10.1038/s41467-018-06636-w. Nat Commun. 2018. PMID: 30279414 Free PMC article.
Abstract
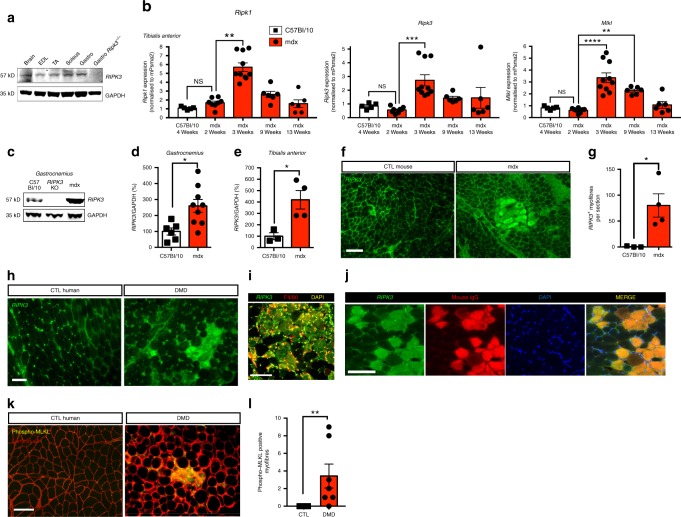
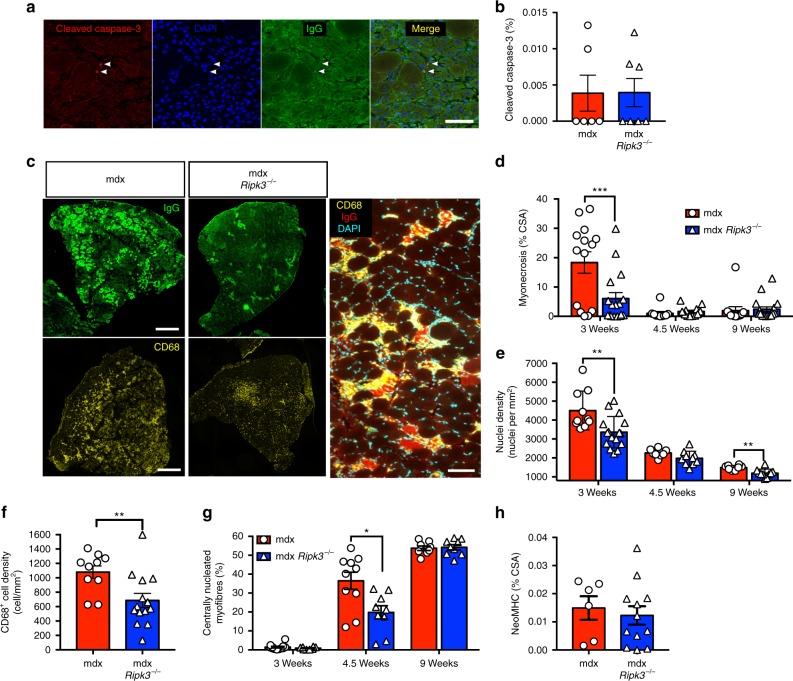
Duchenne muscular dystrophy (DMD) is a severe degenerative disorder caused by mutations in the dystrophin gene. Dystrophin-deficient muscles are characterised by progressive myofibre necrosis in which inflammation plays a deleterious role. However, the molecular mechanisms underlying inflammation-induced necrosis in muscle cells are unknown. Here we show that necroptosis is a mechanism underlying myofibre death in dystrophin-deficient muscle. RIPK1, RIPK3 and MLKL are upregulated in dystrophic mouse myofibres. In human DMD samples, there is strong immunoreactivity to RIPK3 and phospho-MLKL in myofibres. In vitro, TNFα can elicit necroptosis in C2C12 myoblasts, and RIPK3 overexpression sensitises myoblasts to undergo TNF-induced death. Furthermore, genetic ablation of Ripk3 in mdx mice reduces myofibre degeneration, inflammatory infiltrate, and muscle fibrosis, and eventually improves muscle function. These findings provide the first evidence of necroptotic cell death in a disease affecting skeletal muscle and identify RIPK3 as a key player in the degenerative process in dystrophin-deficient muscles.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Tidball JG, Albrecht DE, Lokensgard BE, Spencer MJ. Apoptosis precedes necrosis of dystrophin-deficient muscle. J. Cell Sci. 1995;108(Pt 6):2197–2204. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
