

Amelioration of mitochondrial dysfunction in heart failure through S-sulfhydration of Ca2+/calmodulin-dependent protein kinase II
- PMID: 30195191
- PMCID: PMC6128039
- DOI: 10.1016/j.redox.2018.08.008
Amelioration of mitochondrial dysfunction in heart failure through S-sulfhydration of Ca2+/calmodulin-dependent protein kinase II
Abstract
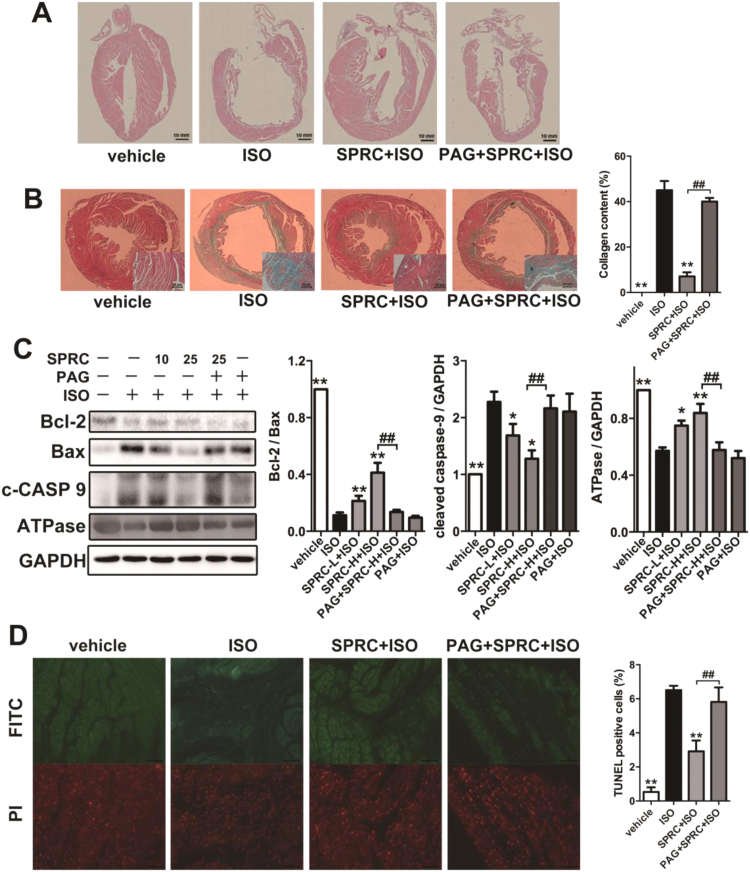
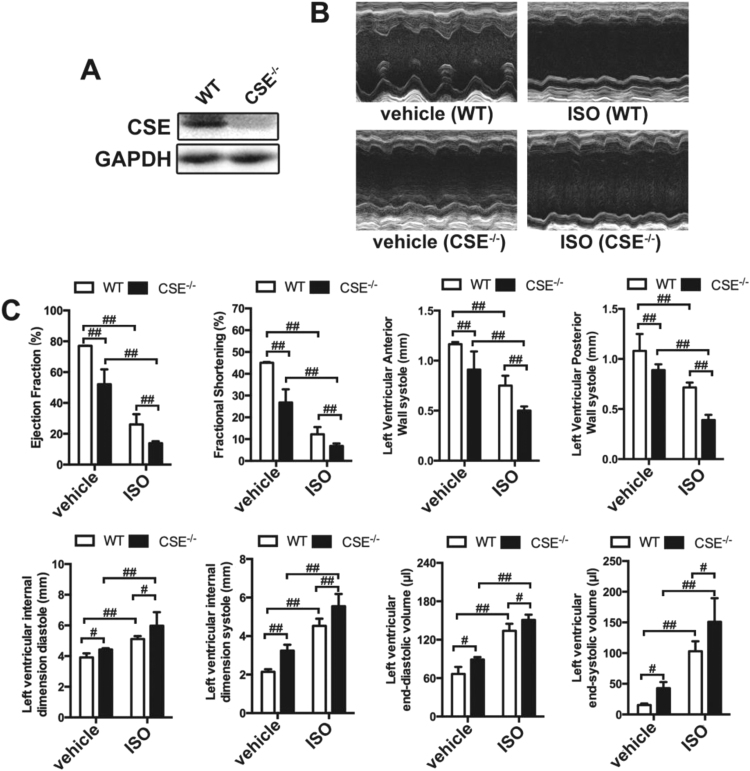
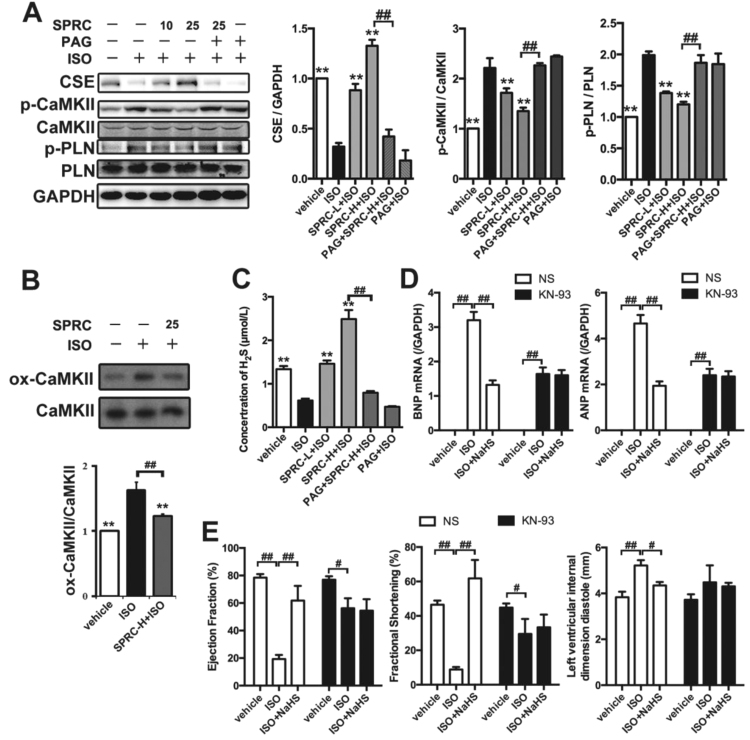
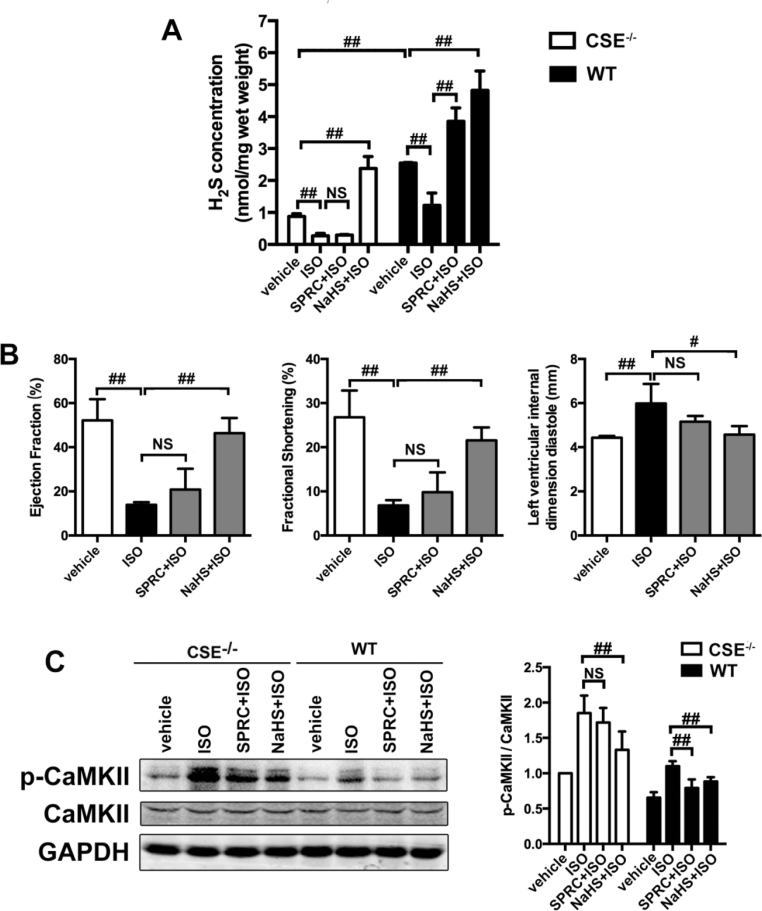
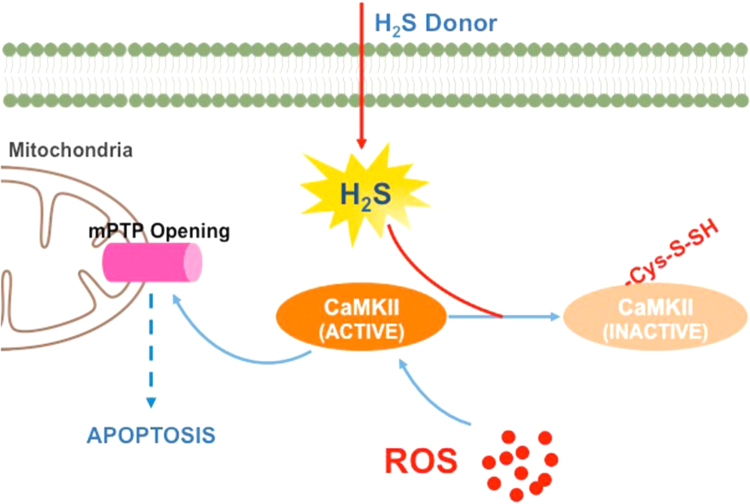
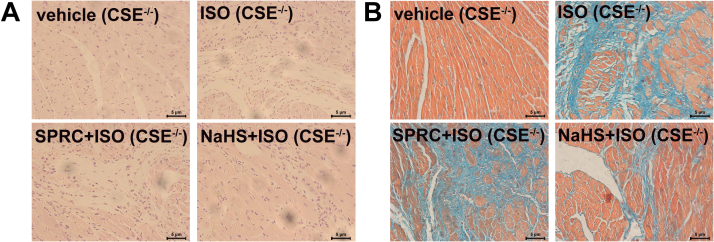
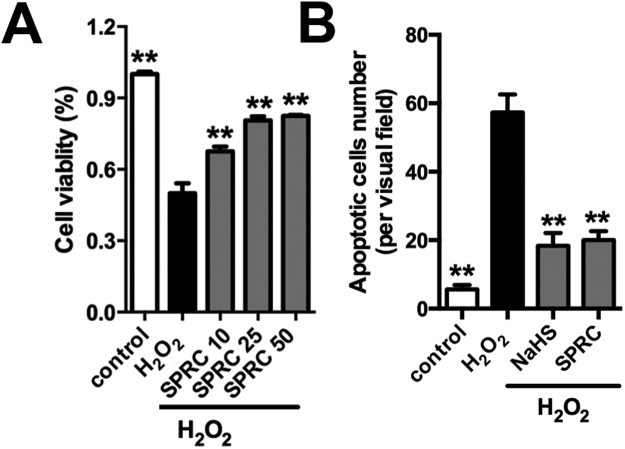
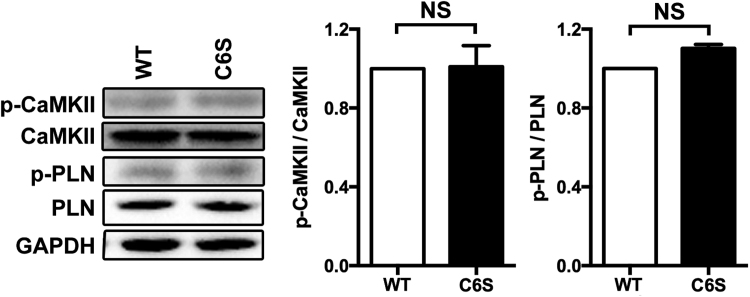
Aims: Ca2+/calmodulin-dependent protein kinase II (CaMKII) plays a critical role in the development of heart failure and in the induction of myocardial mitochondrial injury. Recent evidence has shown that hydrogen sulfide (H2S), produced by the enzyme cystathionine γ-lyase (CSE), improves the cardiac function in heart failure. However, the cellular mechanisms for this remain largely unknown. The present study was conducted to determine the functional role of H2S in protecting against mitochondrial dysfunction in heart failure through the inhibition of CaMKII using wild type and CSE knockout mouse models.
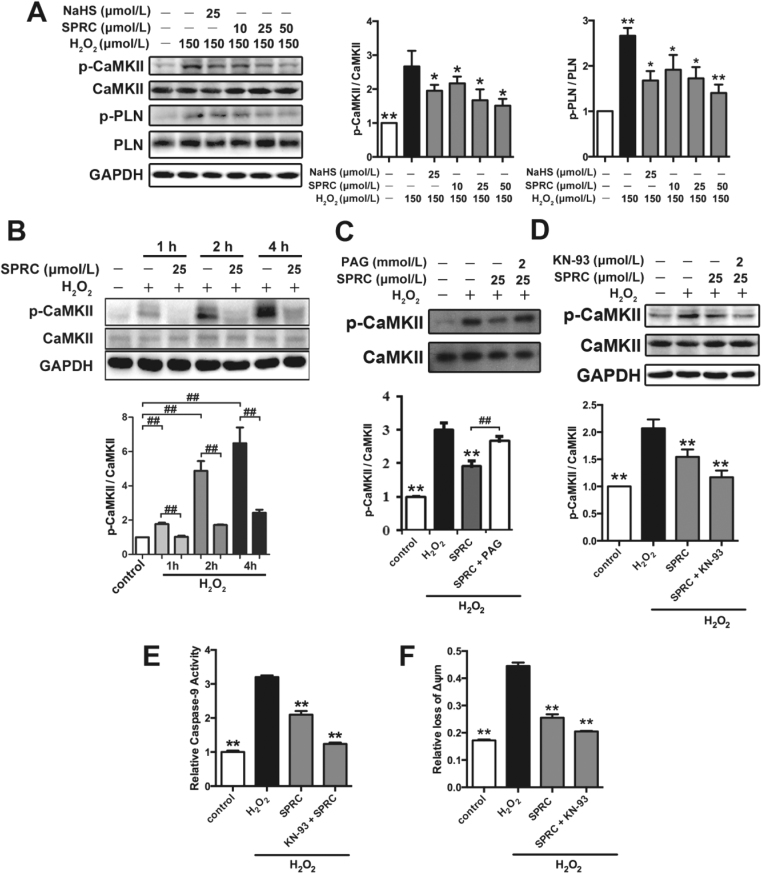
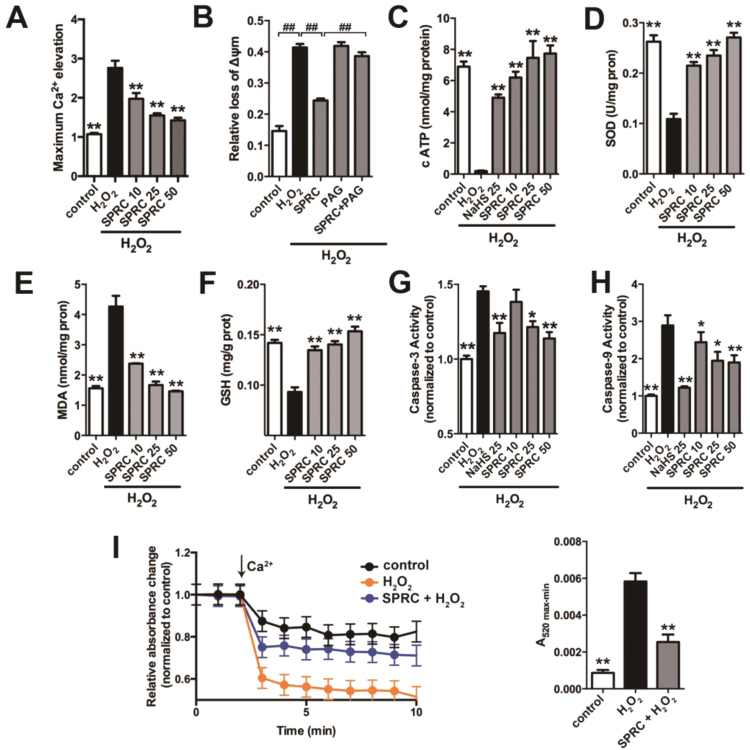
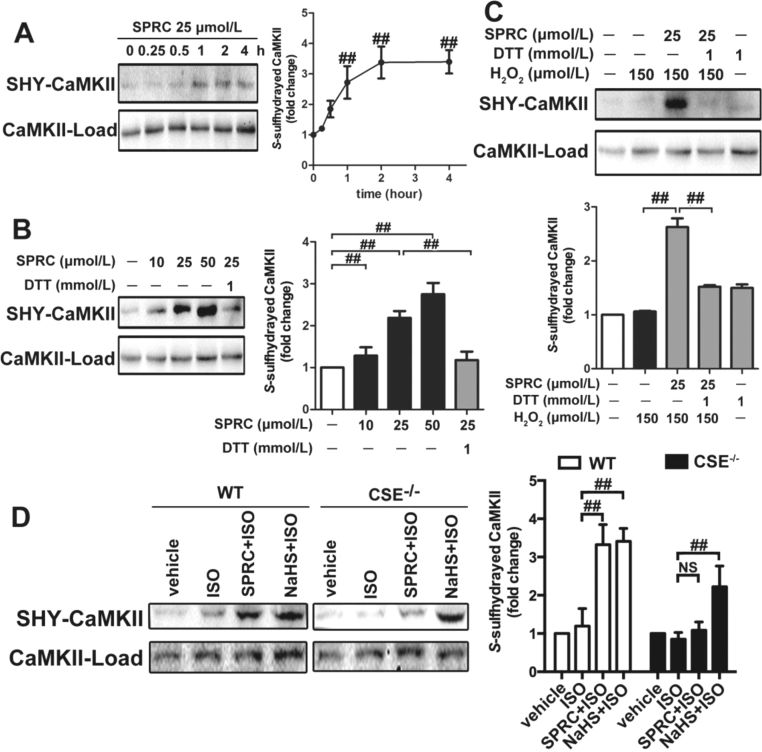
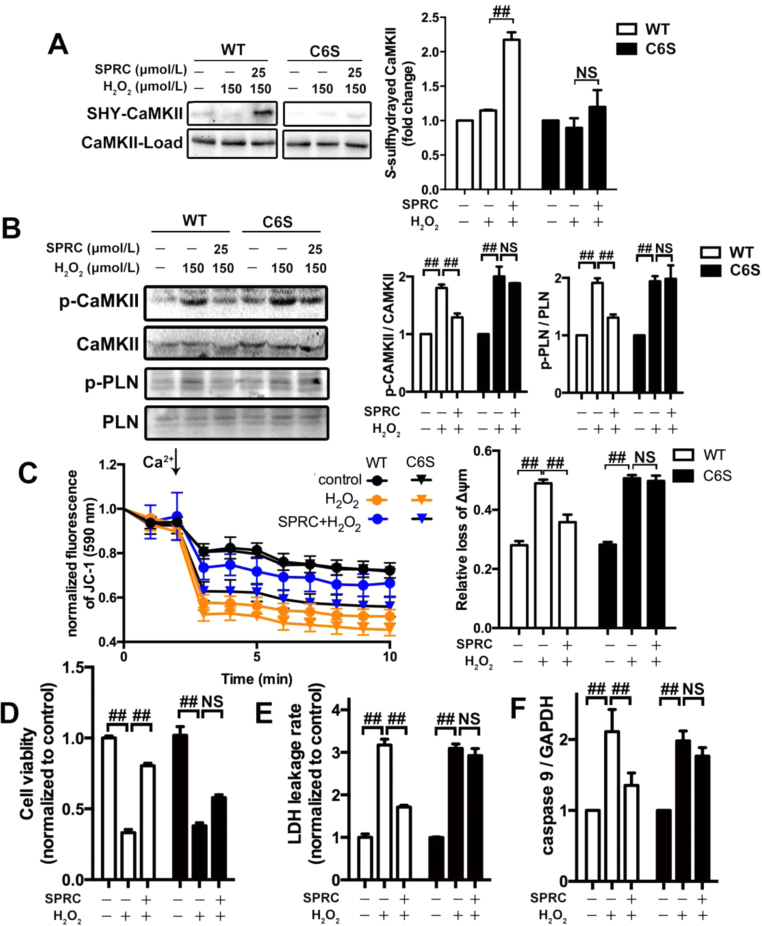
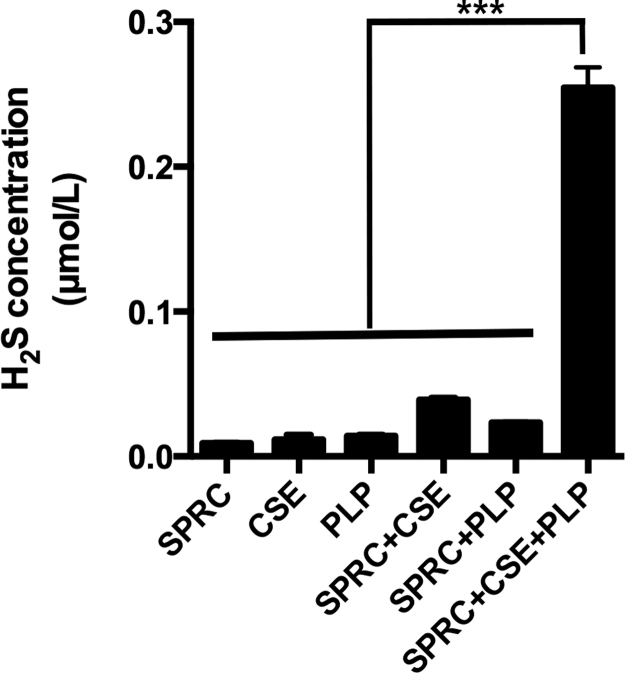
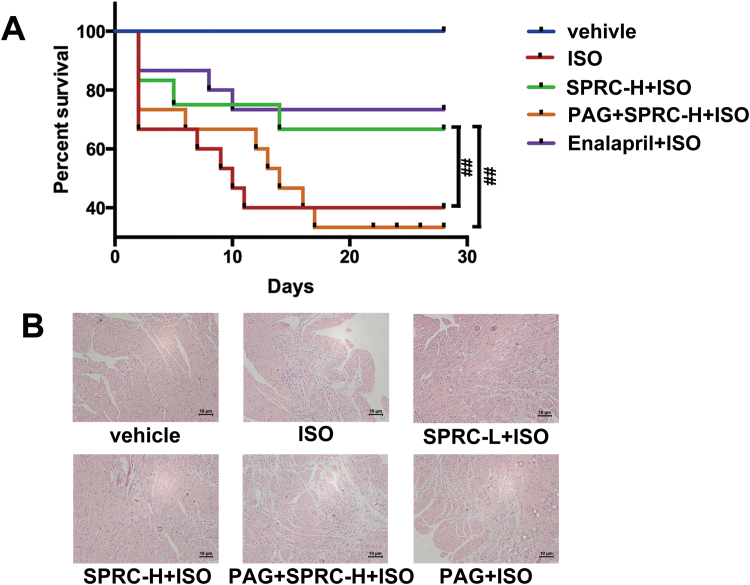
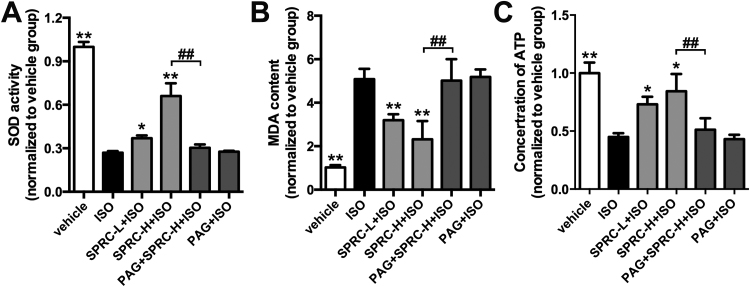
Results: Treatment with S-propyl-L-cysteine (SPRC) or sodium hydrosulfide (NaHS), modulators of blood H2S levels, attenuated the development of heart failure in animals, reduced lipid peroxidation, and preserved mitochondrial function. The inhibition CaMKII phosphorylation by SPRC and NaHS as demonstrated using both in vivo and in vitro models corresponded with the cardioprotective effects of these compounds. Interestingly, CaMKII activity was found to be elevated in CSE knockout (CSE-/-) mice as compared to wild type animals and the phosphorylation status of CaMKII appeared to relate to the severity of heart failure. Importantly, in wild type mice SPRC was found to promote S-sulfhydration of CaMKII leading to reduced activity of this protein, however, in CSE-/- mice S-sulfhydration was abolished following SPRC treatment.
Innovation and conclusions: A novel mechanism depicting a role of S-sulfhydration in the regulation of CaMKII is presented. SPRC mediated S-sulfhydration of CaMKII was found to inhibit CAMKII activity and to preserve cardiovascular homeostasis.
Keywords: Ca(2+)/calmodulin-dependent protein kinase II; Heart Failure; Hydrogen Sulfide; Mitochondria; S-sulfhydration.
Copyright © 2018. Published by Elsevier B.V.
Figures
Similar articles
-
Cystathionine γ Lyase Sulfhydrates the RNA Binding Protein Human Antigen R to Preserve Endothelial Cell Function and Delay Atherogenesis.Circulation. 2019 Jan 2;139(1):101-114. doi: 10.1161/CIRCULATIONAHA.118.034757. Circulation. 2019. PMID: 29970364
-
S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics.Pharmacol Res. 2016 Nov;113(Pt A):116-124. doi: 10.1016/j.phrs.2016.08.023. Epub 2016 Aug 20. Pharmacol Res. 2016. PMID: 27553984 Free PMC article.
-
Cystathionine γ lyase S-sulfhydrates Drp1 to ameliorate heart dysfunction.Redox Biol. 2022 Dec;58:102519. doi: 10.1016/j.redox.2022.102519. Epub 2022 Oct 28. Redox Biol. 2022. PMID: 36327794 Free PMC article.
-
Investigating the impact of protein S-sulfhydration modification on vascular diseases: A comprehensive review.Eur J Pharmacol. 2024 Mar 5;966:176345. doi: 10.1016/j.ejphar.2024.176345. Epub 2024 Jan 19. Eur J Pharmacol. 2024. PMID: 38244760 Review.
-
CaMKII is a nodal signal for multiple programmed cell death pathways in heart.J Mol Cell Cardiol. 2017 Feb;103:102-109. doi: 10.1016/j.yjmcc.2016.12.007. Epub 2016 Dec 24. J Mol Cell Cardiol. 2017. PMID: 28025046 Free PMC article. Review.
Cited by
-
New Targets in Heart Failure Drug Therapy.Front Cardiovasc Med. 2021 May 5;8:665797. doi: 10.3389/fcvm.2021.665797. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 34026873 Free PMC article. Review.
-
Sulfur signaling pathway in cardiovascular disease.Front Pharmacol. 2023 Nov 24;14:1303465. doi: 10.3389/fphar.2023.1303465. eCollection 2023. Front Pharmacol. 2023. PMID: 38074127 Free PMC article. Review.
-
Effect of Sodium Thiosulfate Pre-Treatment on Renal Ischemia-Reperfusion Injury in Kidney Transplantation.Int J Mol Sci. 2024 Sep 2;25(17):9529. doi: 10.3390/ijms25179529. Int J Mol Sci. 2024. PMID: 39273476 Free PMC article.
-
Persulfidation of transcription factor FOXO1 at cysteine 457: A novel mechanism by which H2S inhibits vascular smooth muscle cell proliferation.J Adv Res. 2020 Jul 1;27:155-164. doi: 10.1016/j.jare.2020.06.023. eCollection 2021 Jan. J Adv Res. 2020. PMID: 33318874 Free PMC article. Review.
-
S-Propargyl-Cysteine Attenuates Stroke Heterogeneity via Promoting Protective Autophagy Across Multiple Neural Cell Types: Insights From Single-Cell Sequencing.CNS Neurosci Ther. 2025 Jul;31(7):e70399. doi: 10.1111/cns.70399. CNS Neurosci Ther. 2025. PMID: 40702904 Free PMC article.
References
-
- Altaany Z., Ju Y., Yang G., Wang R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci. Signal. 2014;7:ra87. - PubMed
-
- Ariely R., Evans K., Mills T. Heart failure in China: a review of the literature. Drugs. 2013;73:689–701. - PubMed
-
- Atkins B.Z., Hashmi Z.A., Ganapathi A.M., Harrison J.K., Hughes G.C., Rogers J.G., Milano C.A. Surgical correction of aortic valve insufficiency after left ventricular assist device implantation. J. Thorac. Cardiovasc. Surg. 2013;146:1247–1252. - PubMed
-
- Bir S.C., Kolluru G.K., McCarthy P., Shen X., Pardue S., Pattillo C.B., Kevil C.G. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis. J. Am. Heart Assoc. 2012;1:e004093. - PMC - PubMed
-
- Coletta C., Papapetropoulos A., Erdelyi K., Olah G., Modis K., Panopoulos P., Asimakopoulou A., Gero D., Sharina I., Martin E., Szabo C. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA. 2012;109:9161–9166. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
