

Machine Learning Predicts the Yeast Metabolome from the Quantitative Proteome of Kinase Knockouts
- PMID: 30195436
- PMCID: PMC6167078
- DOI: 10.1016/j.cels.2018.08.001
Machine Learning Predicts the Yeast Metabolome from the Quantitative Proteome of Kinase Knockouts
Abstract
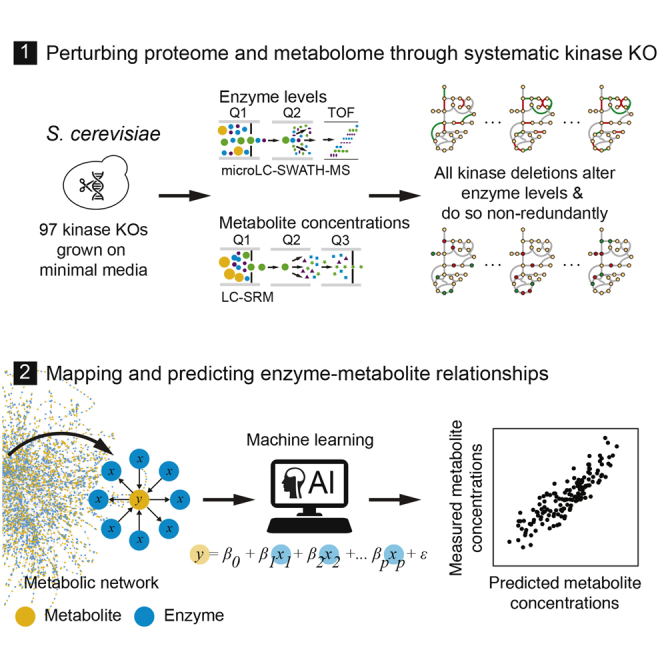
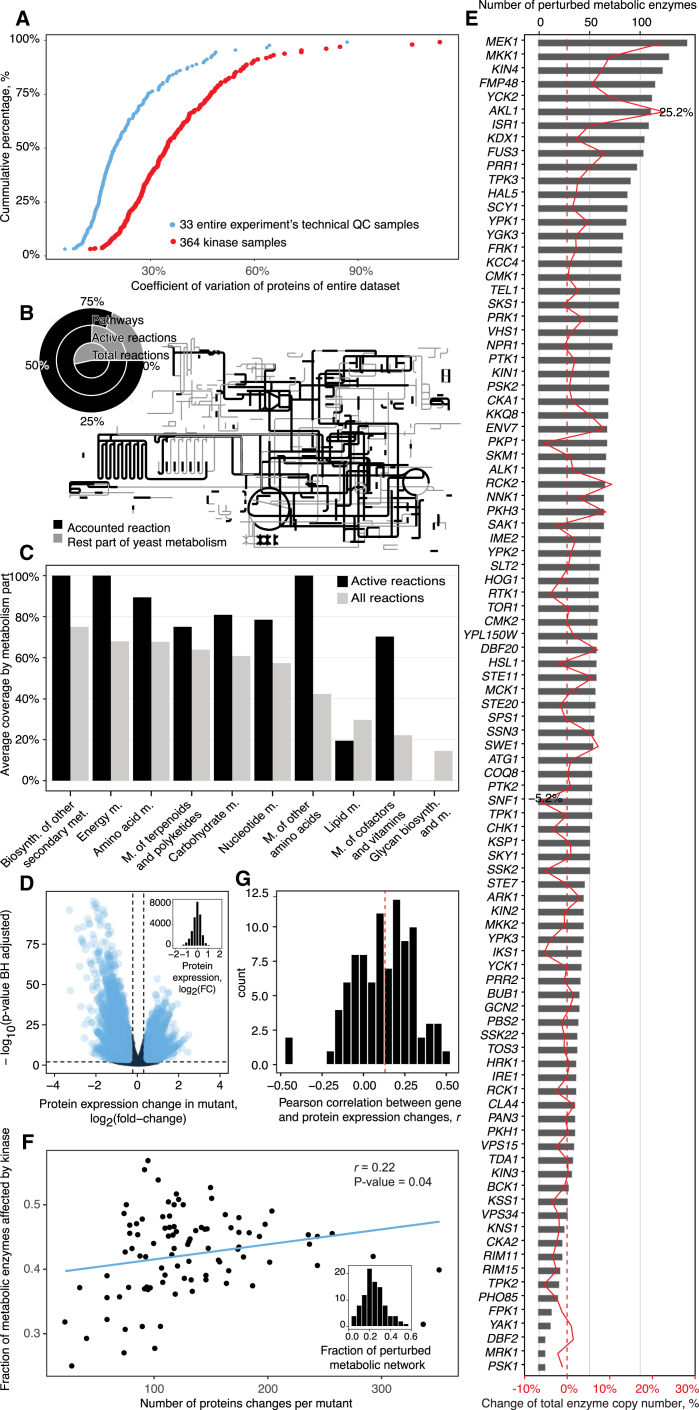
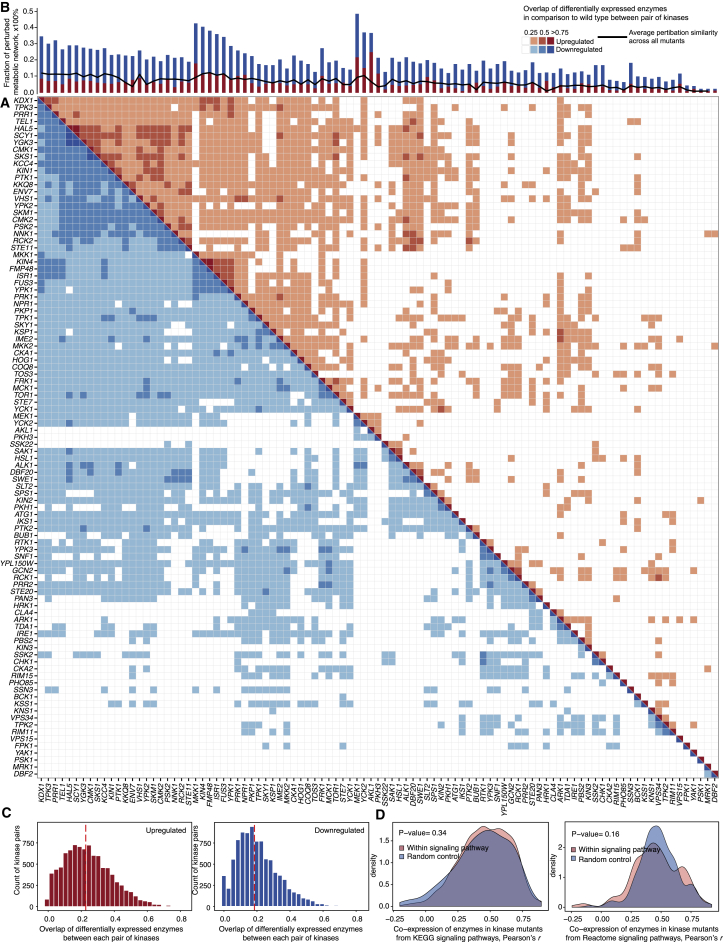
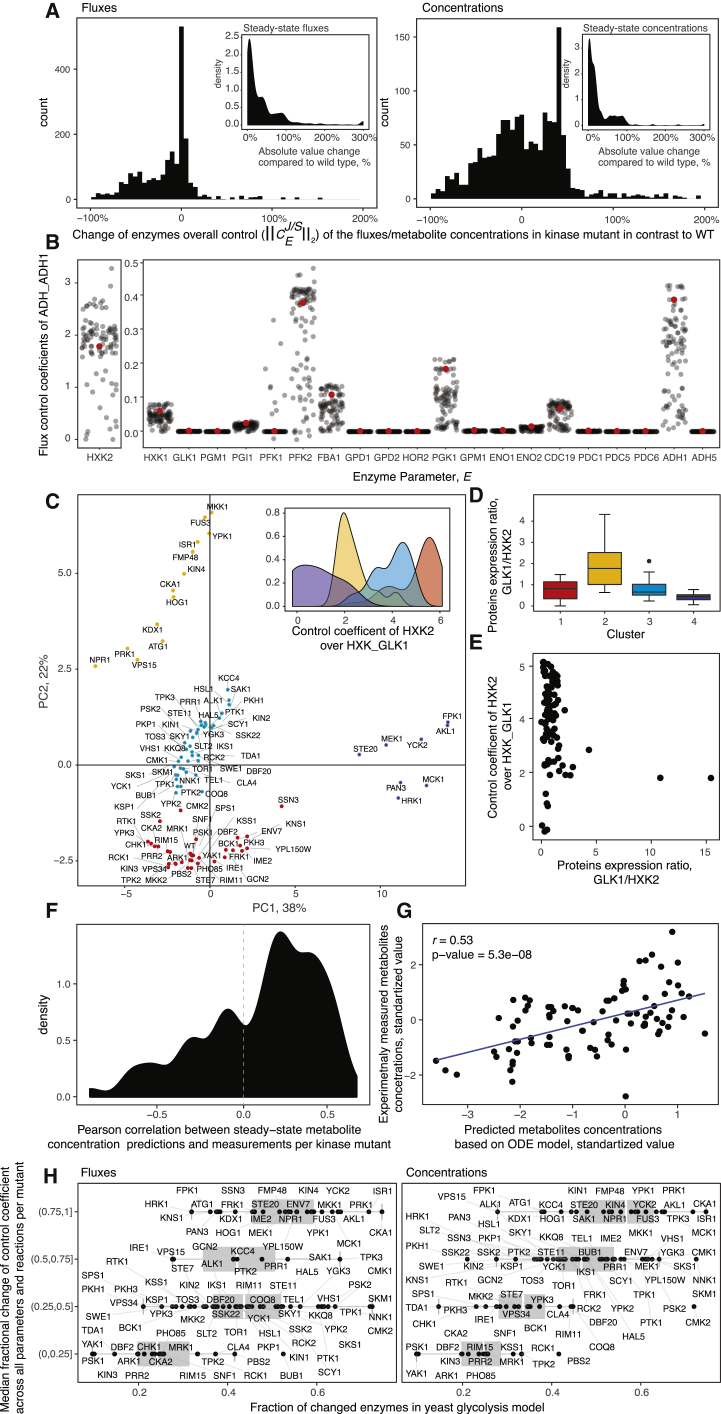
A challenge in solving the genotype-to-phenotype relationship is to predict a cell's metabolome, believed to correlate poorly with gene expression. Using comparative quantitative proteomics, we found that differential protein expression in 97 Saccharomyces cerevisiae kinase deletion strains is non-redundant and dominated by abundance changes in metabolic enzymes. Associating differential enzyme expression landscapes to corresponding metabolomes using network models provided reasoning for poor proteome-metabolome correlations; differential protein expression redistributes flux control between many enzymes acting in concert, a mechanism not captured by one-to-one correlation statistics. Mapping these regulatory patterns using machine learning enabled the prediction of metabolite concentrations, as well as identification of candidate genes important for the regulation of metabolism. Overall, our study reveals that a large part of metabolism regulation is explained through coordinated enzyme expression changes. Our quantitative data indicate that this mechanism explains more than half of metabolism regulation and underlies the interdependency between enzyme levels and metabolism, which renders the metabolome a predictable phenotype.
Keywords: enzyme abundance; genotype-phenotype problem; hierarchical regulation; high-throughput proteomics; machine learning; metabolic control analysis; metabolism; multi-omics.
Copyright © 2018 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
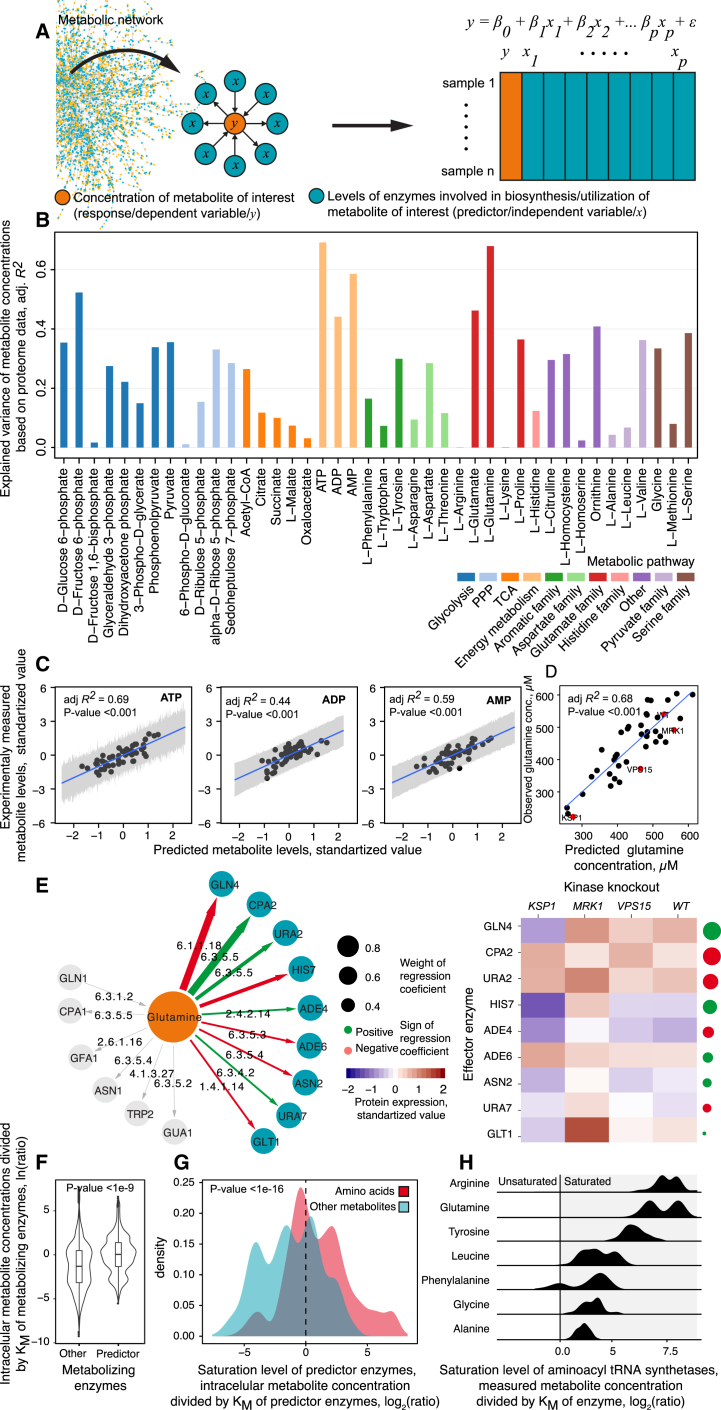
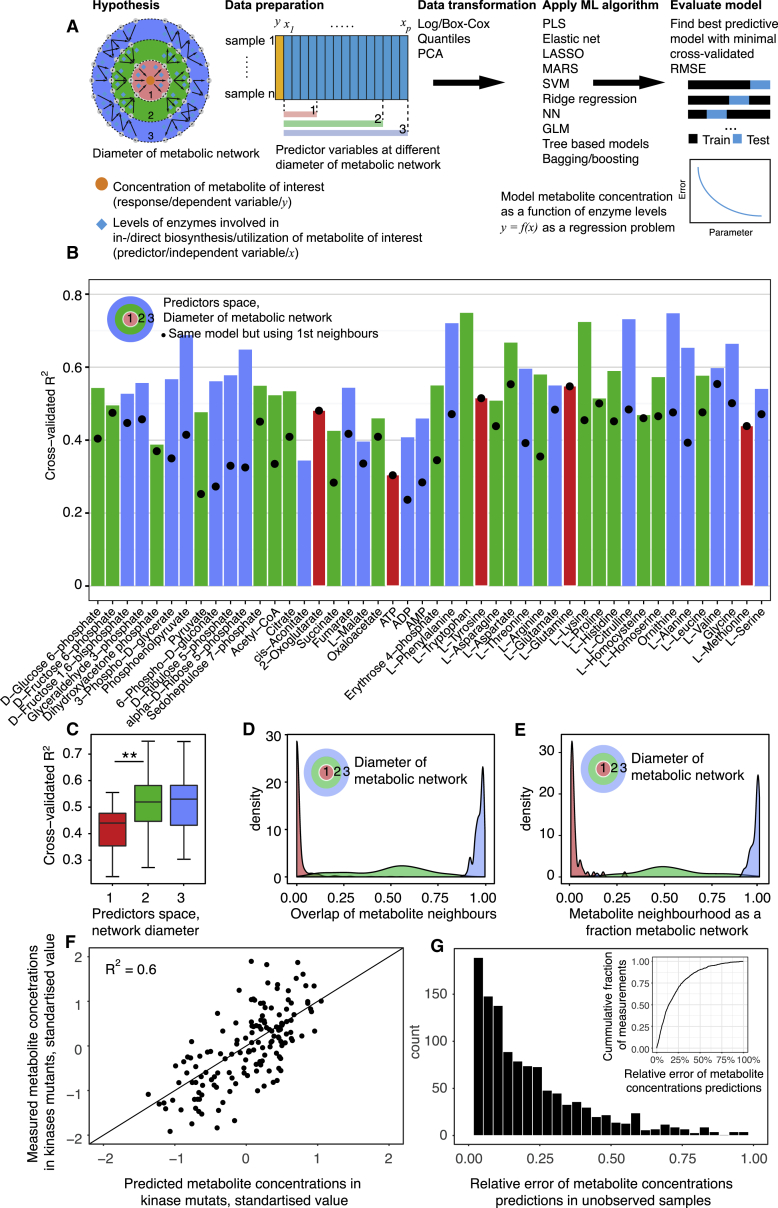
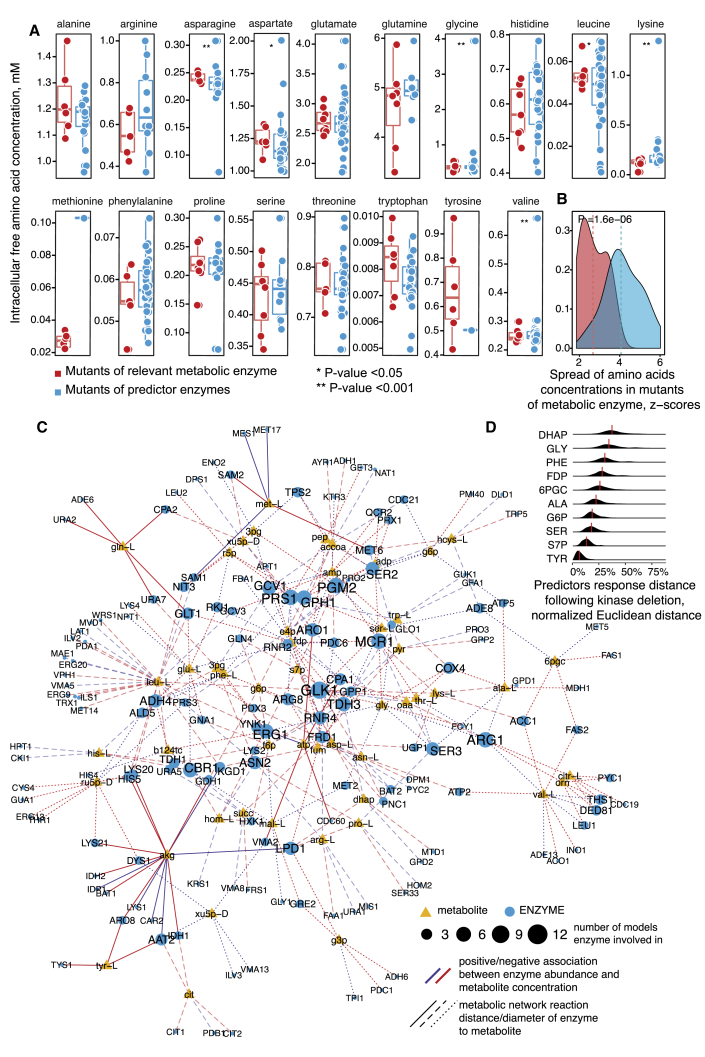
References
-
- Akaike H. A new look at the statistical model identification. IEEE Trans. Automat. Contr. 1974;19:716–723.
-
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. Stat. Methodol. 1995;57:289–300.
-
- Beyenbach K.W., Wieczorek H. The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J. Exp. Biol. 2006;209:577–589. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
