

New tools for the analysis and validation of cryo-EM maps and atomic models
- PMID: 30198894
- PMCID: PMC6130467
- DOI: 10.1107/S2059798318009324
New tools for the analysis and validation of cryo-EM maps and atomic models
Abstract
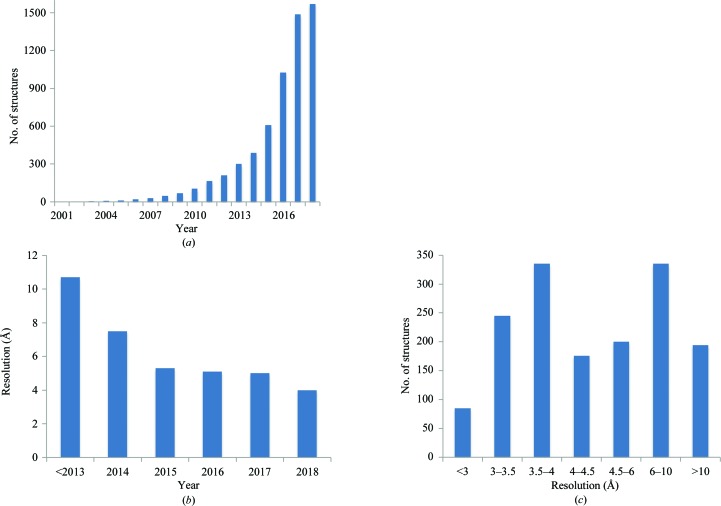
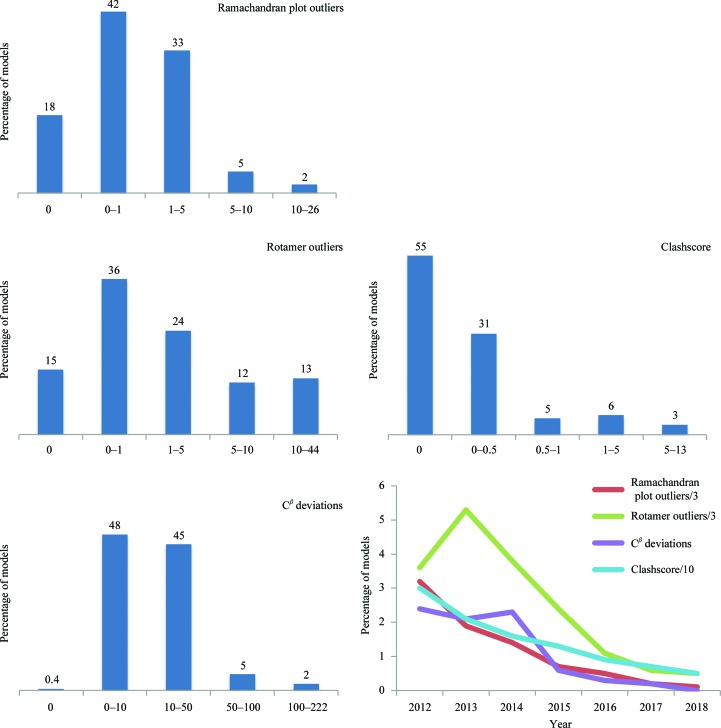
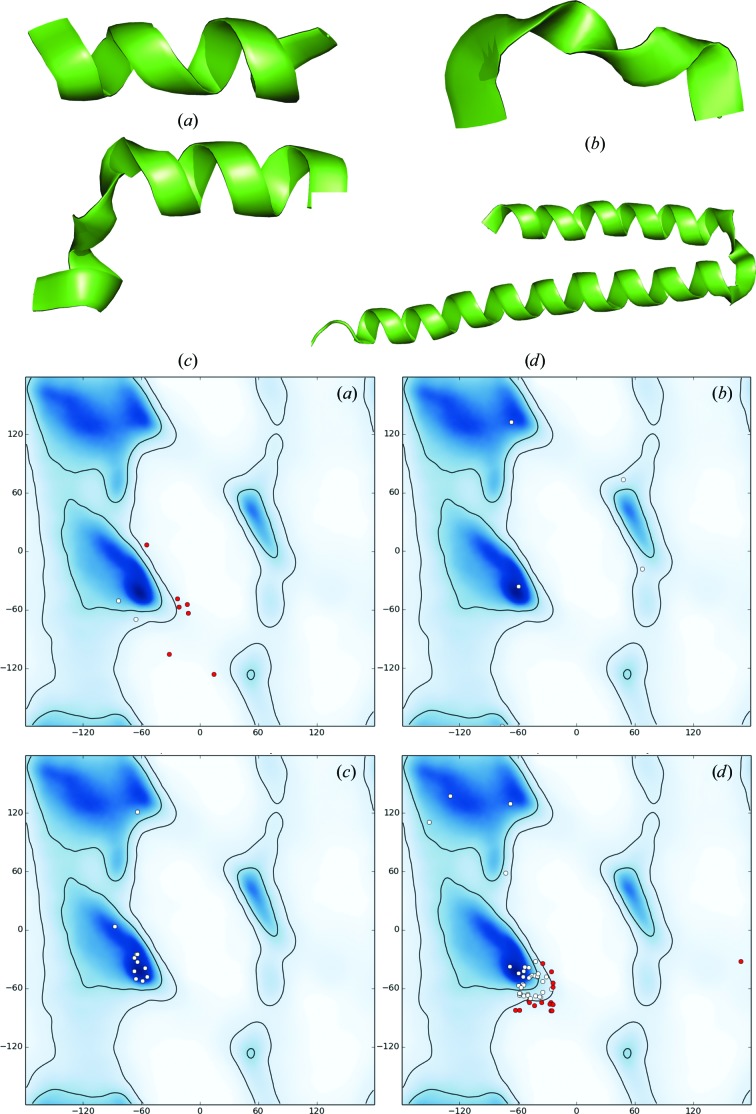
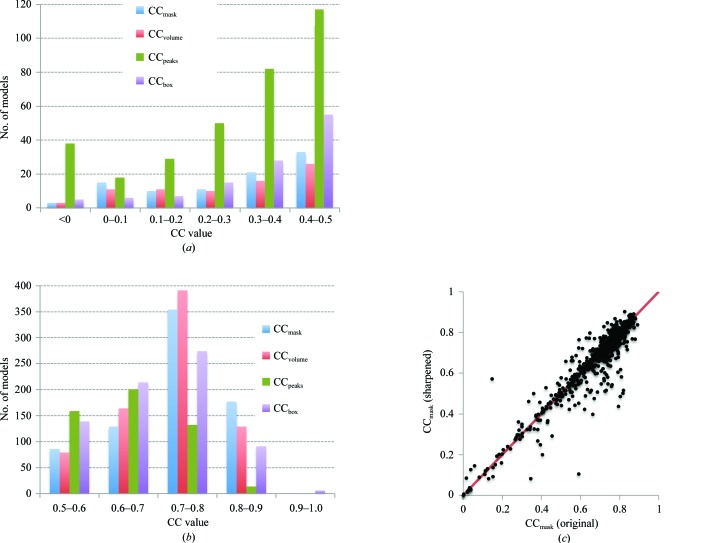
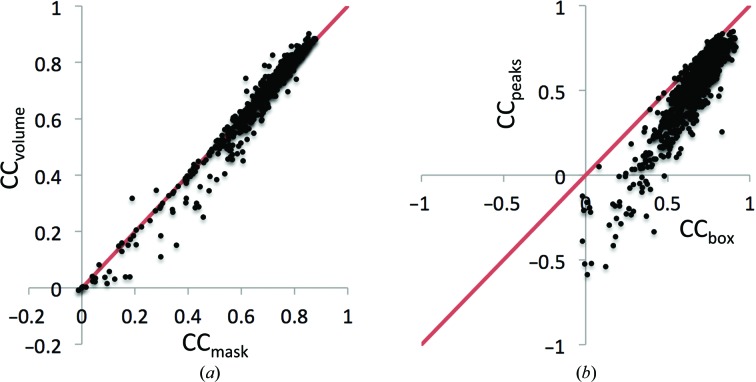
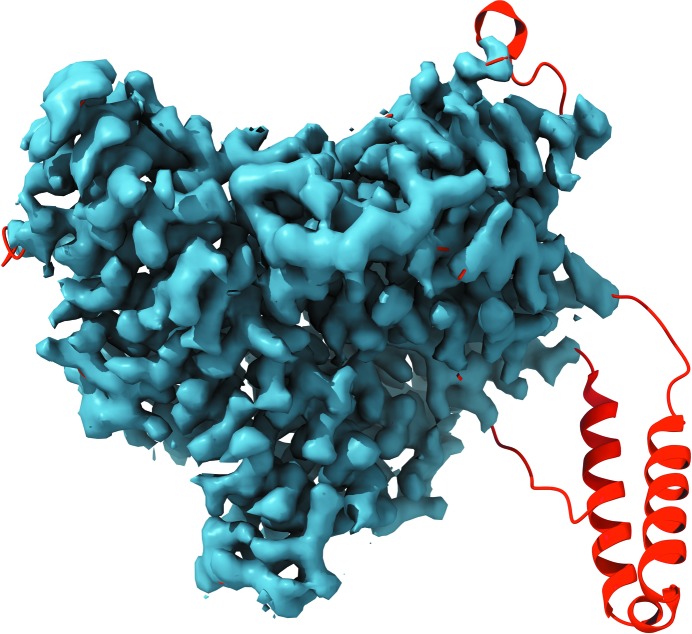
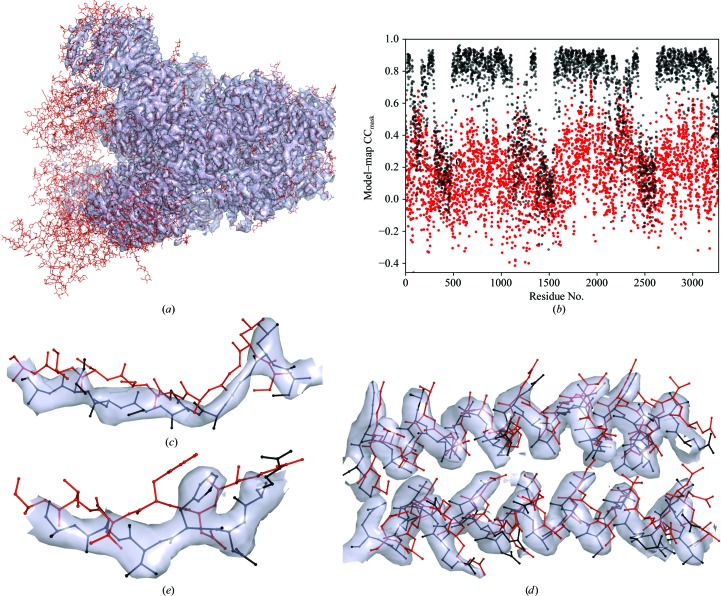
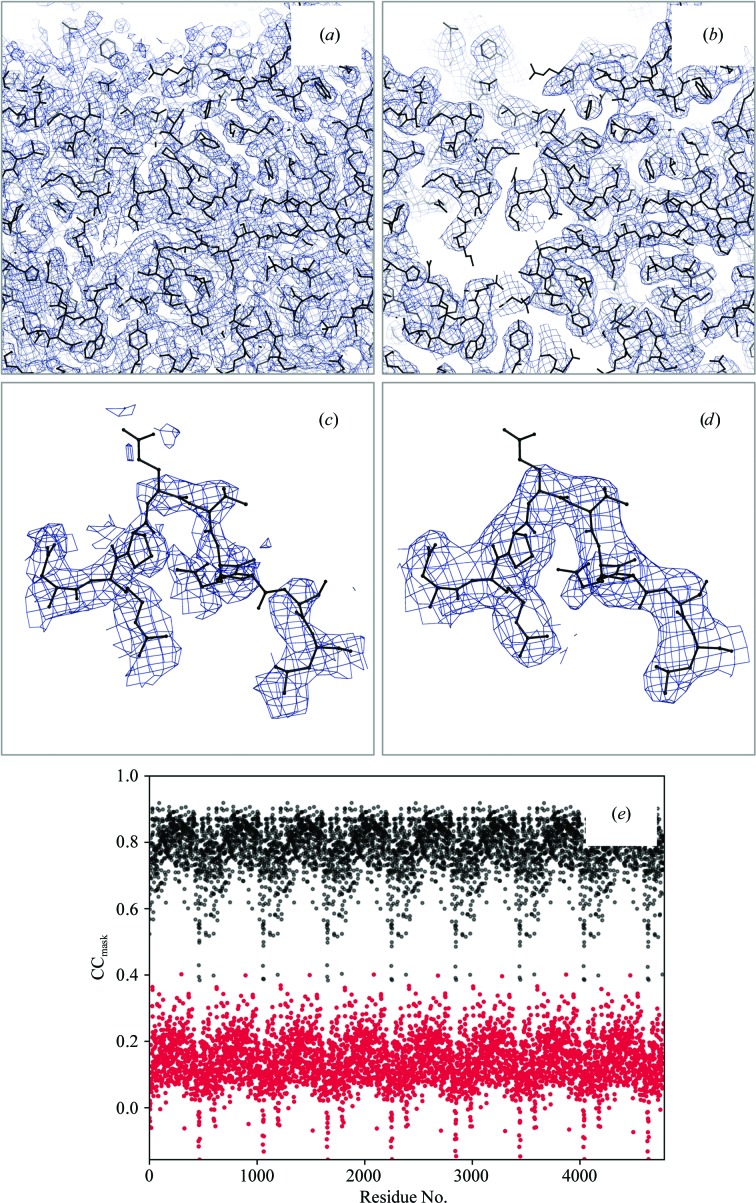
Recent advances in the field of electron cryomicroscopy (cryo-EM) have resulted in a rapidly increasing number of atomic models of biomacromolecules that have been solved using this technique and deposited in the Protein Data Bank and the Electron Microscopy Data Bank. Similar to macromolecular crystallography, validation tools for these models and maps are required. While some of these validation tools may be borrowed from crystallography, new methods specifically designed for cryo-EM validation are required. Here, new computational methods and tools implemented in PHENIX are discussed, including d99 to estimate resolution, phenix.auto_sharpen to improve maps and phenix.mtriage to analyze cryo-EM maps. It is suggested that cryo-EM half-maps and masks should be deposited to facilitate the evaluation and validation of cryo-EM-derived atomic models and maps. The application of these tools to deposited cryo-EM atomic models and maps is also presented.
Keywords: atomic models; cryo-EM; data quality; model quality; resolution; validation.
open access.
Figures
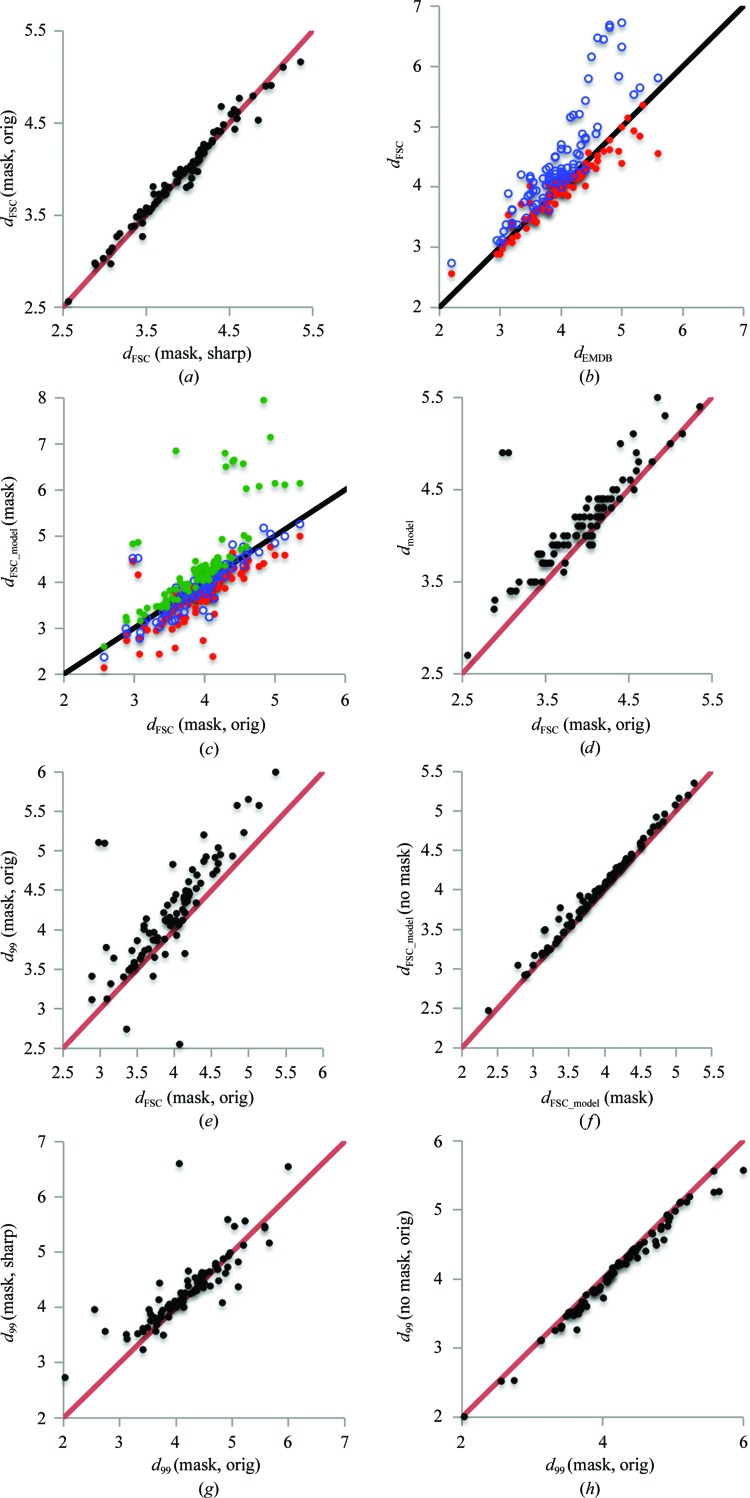
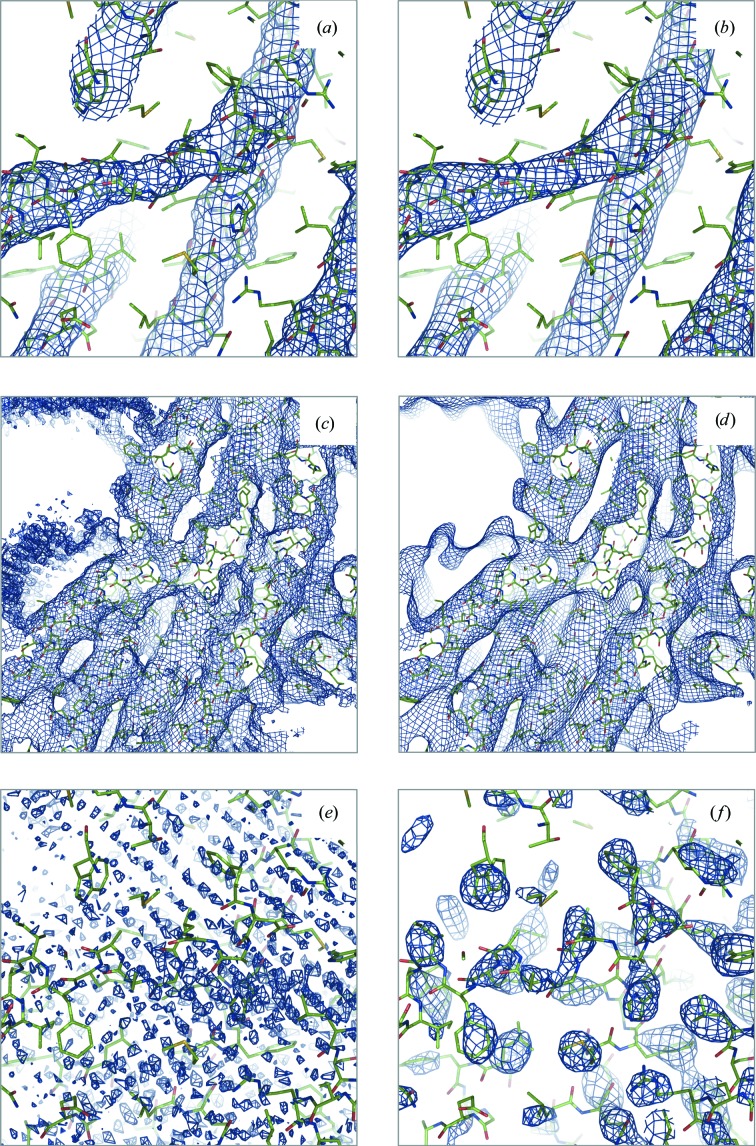
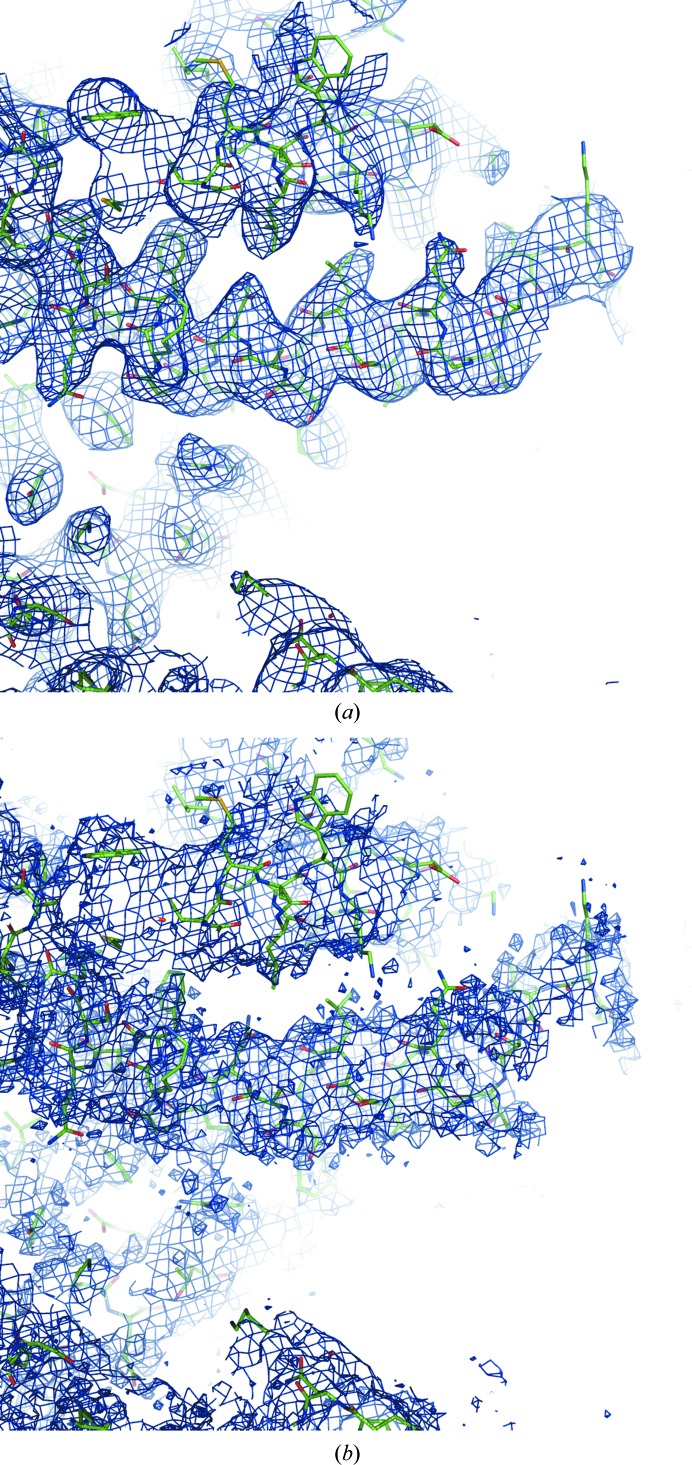
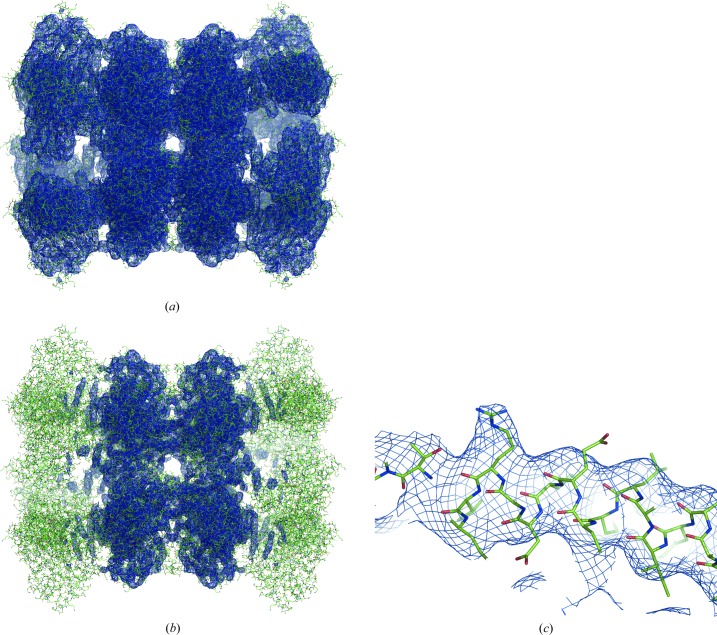
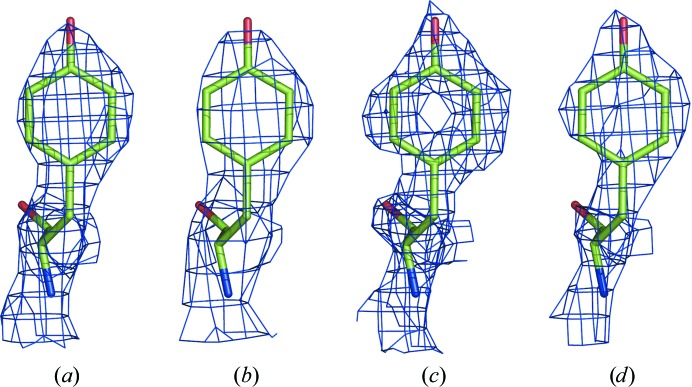
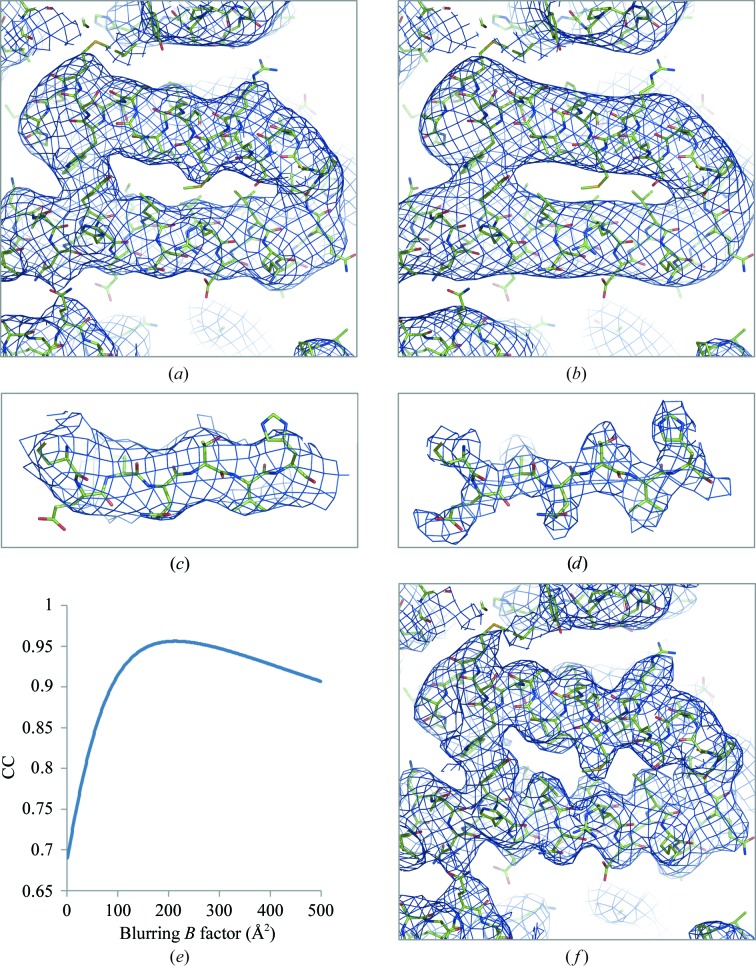
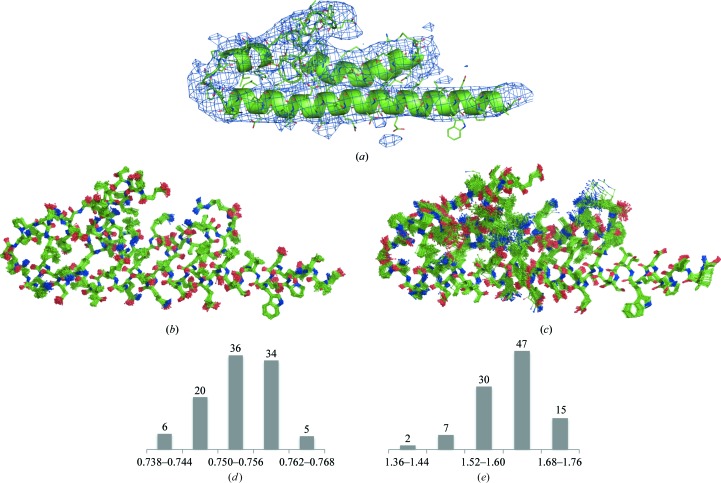
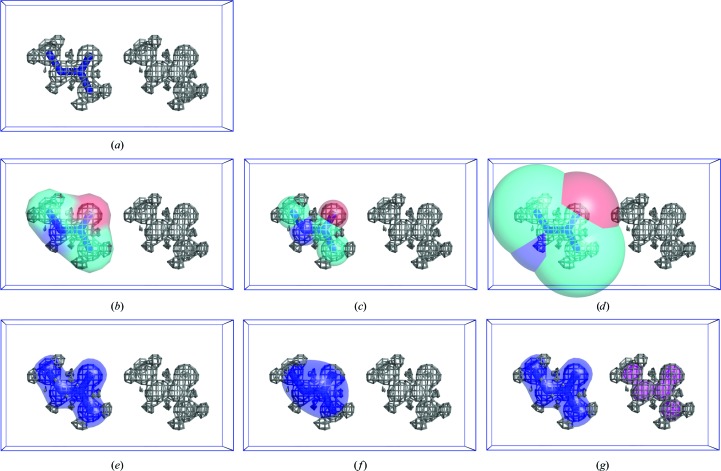
References
-
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221. - PubMed
-
- Afonine, P. V., Headd, J. J., Terwilliger, T. C. & Adams, P. D. (2013). Comput. Crystallogr. Newsl. 4, 43–44.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
