

Copper-dependent amino oxidase 3 governs selection of metabolic fuels in adipocytes
- PMID: 30199530
- PMCID: PMC6130853
- DOI: 10.1371/journal.pbio.2006519
Copper-dependent amino oxidase 3 governs selection of metabolic fuels in adipocytes
Abstract
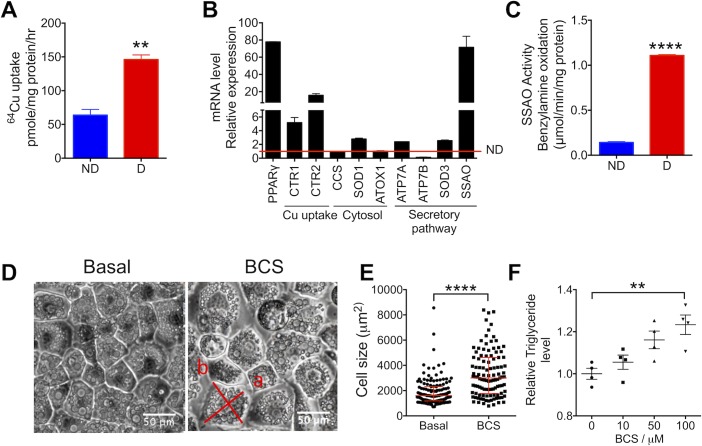
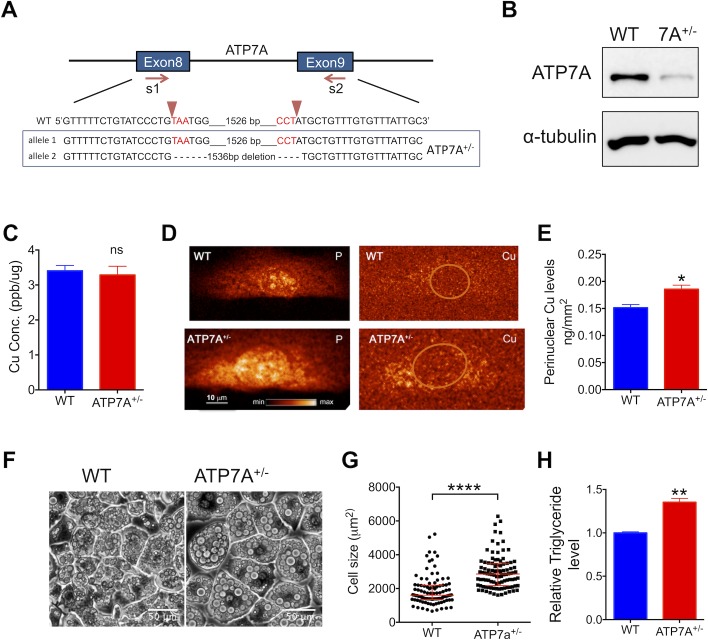
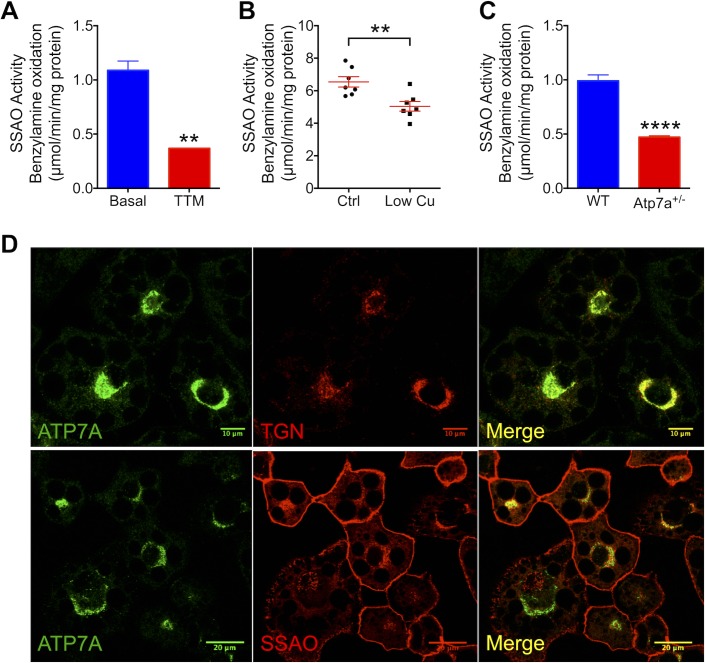
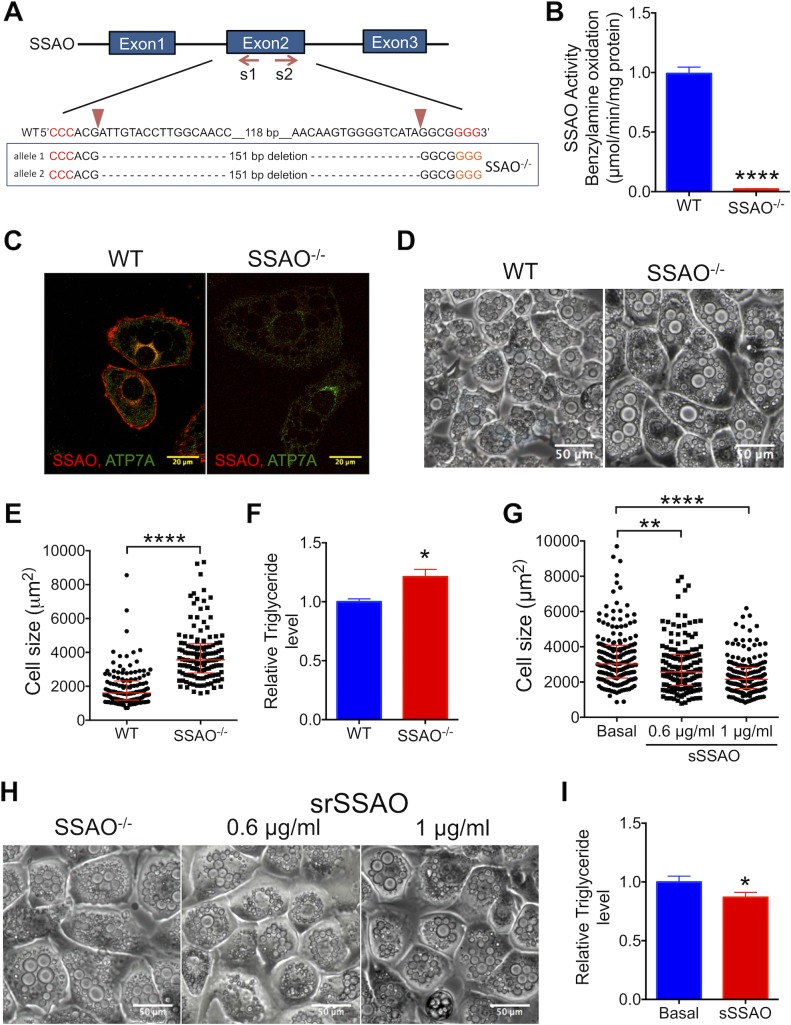
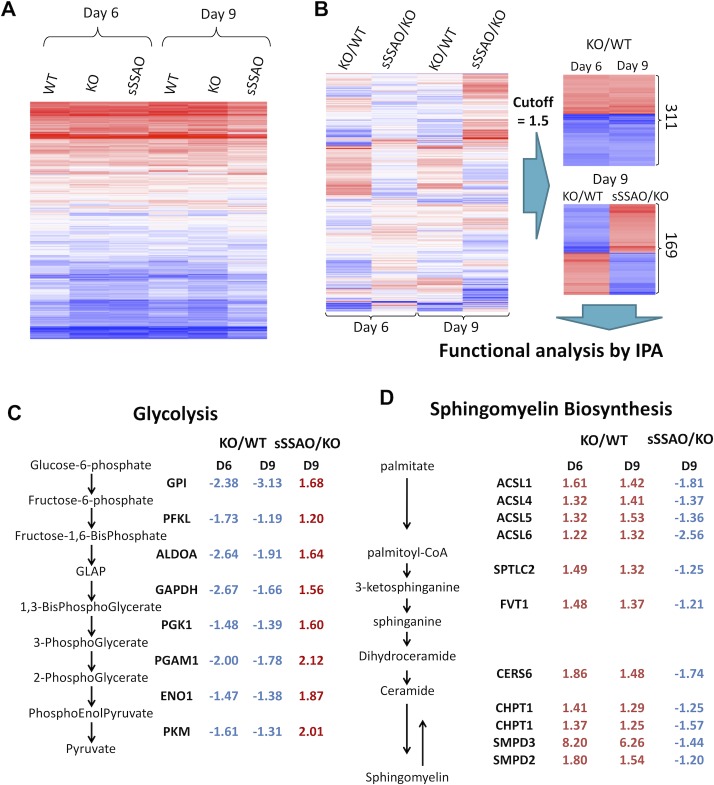
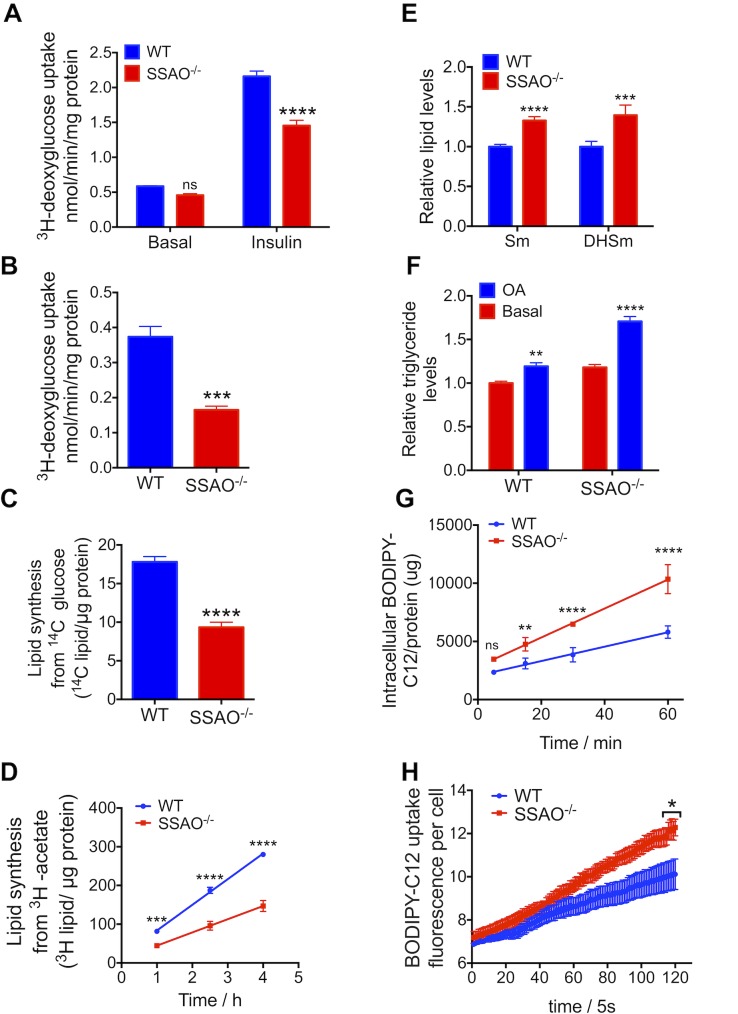
Copper (Cu) has emerged as an important modifier of body lipid metabolism. However, how Cu contributes to the physiology of fat cells remains largely unknown. We found that adipocytes require Cu to establish a balance between main metabolic fuels. Differentiating adipocytes increase their Cu uptake along with the ATP7A-dependent transport of Cu into the secretory pathway to activate a highly up-regulated amino-oxidase copper-containing 3 (AOC3)/semicarbazide-sensitive amine oxidase (SSAO); in vivo, the activity of SSAO depends on the organism's Cu status. Activated SSAO oppositely regulates uptake of glucose and long-chain fatty acids and remodels the cellular proteome to coordinate changes in fuel availability and related downstream processes, such as glycolysis, de novo lipogenesis, and sphingomyelin/ceramide synthesis. The loss of SSAO-dependent regulation due to Cu deficiency, limited Cu transport to the secretory pathway, or SSAO inactivation shifts metabolism towards lipid-dependent pathways and results in adipocyte hypertrophy and fat accumulation. The results establish a role for Cu homeostasis in adipocyte metabolism and identify SSAO as a regulator of energy utilization processes in adipocytes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Kodama H, Murata Y, Kobayashi M. Clinical manifestations and treatment of Menkes disease and its variants. Pediatr Int. 1999;41(4):423–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
