

Description and initial characterization of metatranscriptomic nidovirus-like genomes from the proposed new family Abyssoviridae, and from a sister group to the Coronavirinae, the proposed genus Alphaletovirus
- PMID: 30199753
- PMCID: PMC7112036
- DOI: 10.1016/j.virol.2018.08.010
Description and initial characterization of metatranscriptomic nidovirus-like genomes from the proposed new family Abyssoviridae, and from a sister group to the Coronavirinae, the proposed genus Alphaletovirus
Abstract
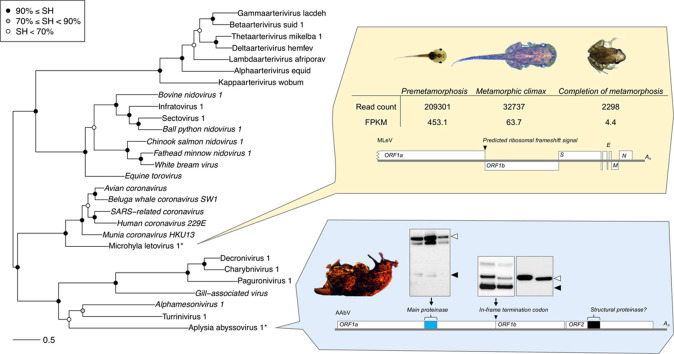
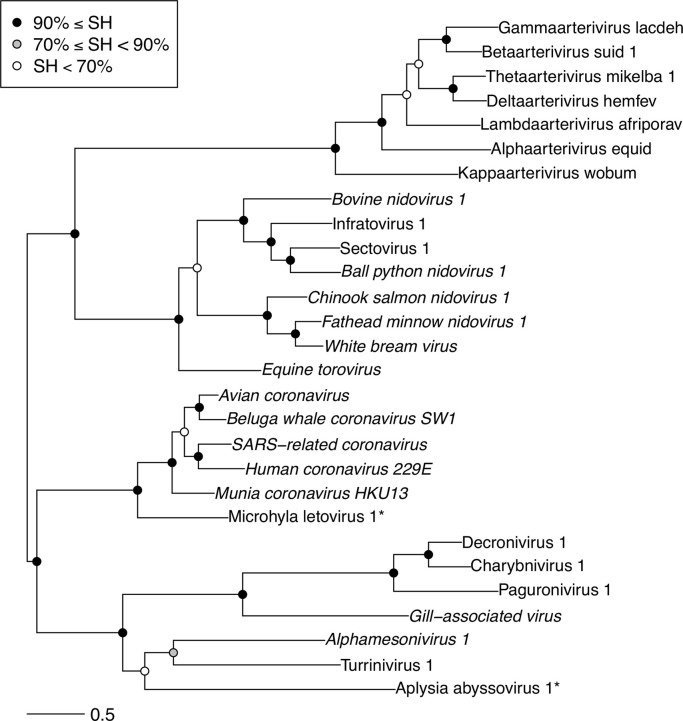
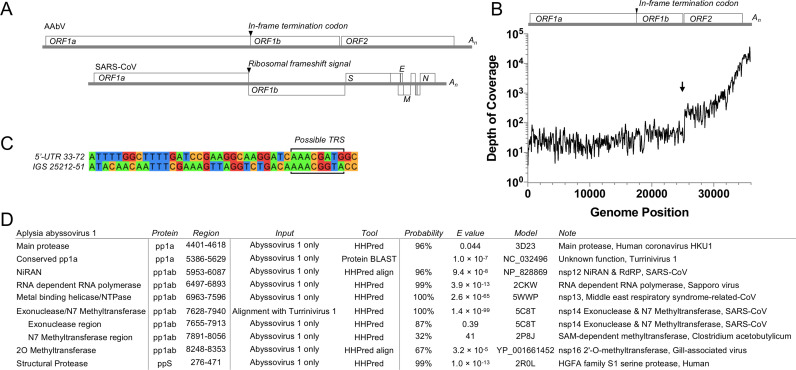
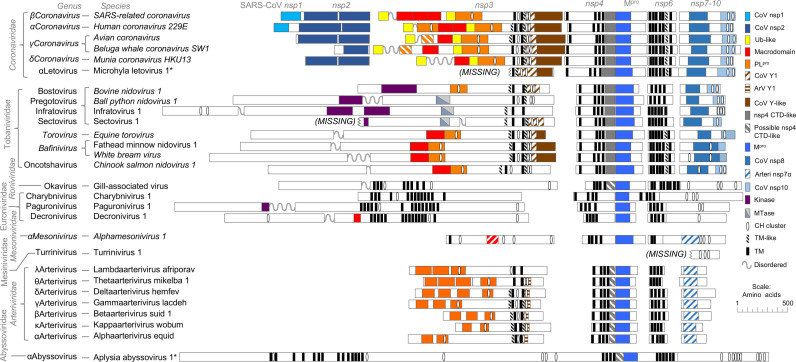
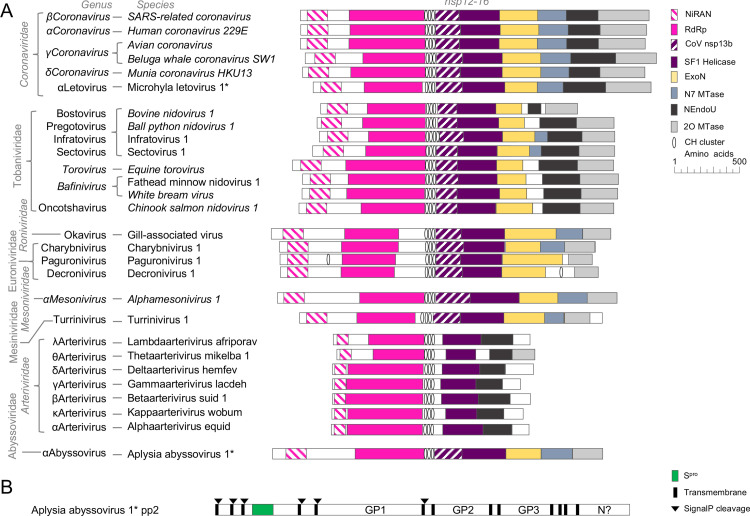
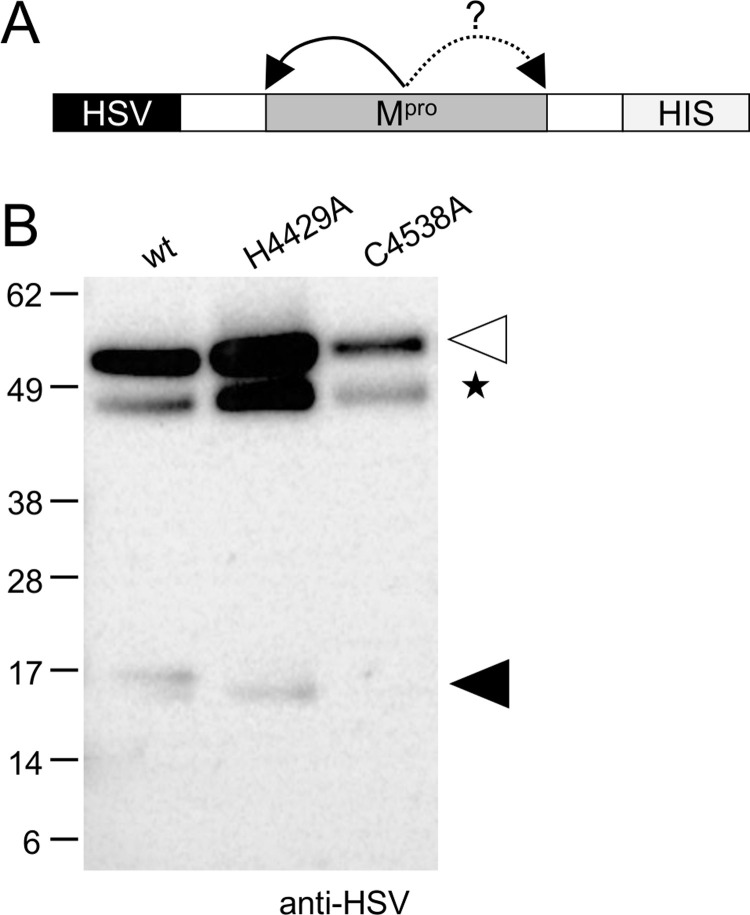
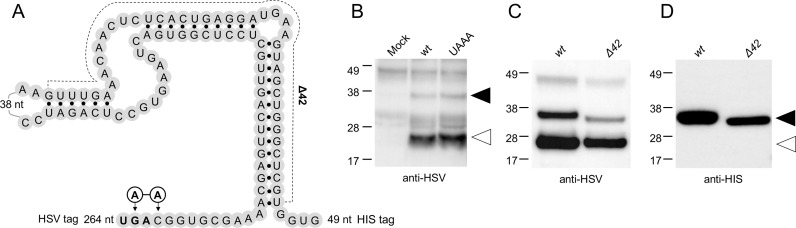
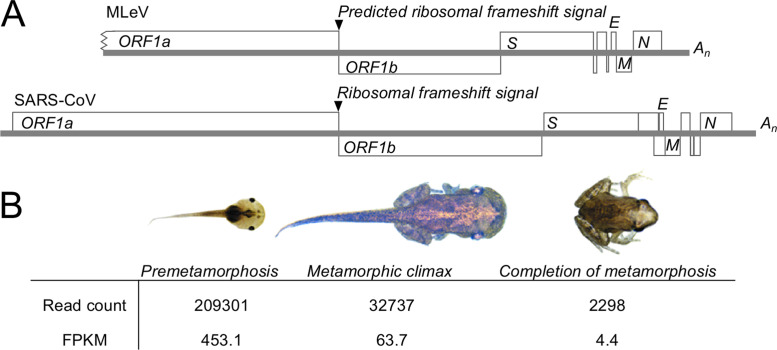
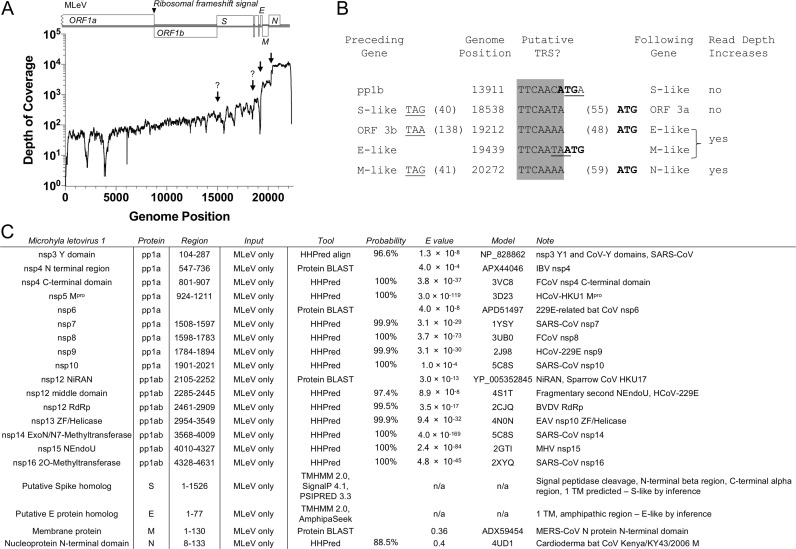
Transcriptomics has the potential to discover new RNA virus genomes by sequencing total intracellular RNA pools. In this study, we have searched publicly available transcriptomes for sequences similar to viruses of the Nidovirales order. We report two potential nidovirus genomes, a highly divergent 35.9 kb likely complete genome from the California sea hare Aplysia californica, which we assign to a nidovirus named Aplysia abyssovirus 1 (AAbV), and a coronavirus-like 22.3 kb partial genome from the ornamented pygmy frog Microhyla fissipes, which we assign to a nidovirus named Microhyla alphaletovirus 1 (MLeV). AAbV was shown to encode a functional main proteinase, and a translational readthrough signal. Phylogenetic analysis suggested that AAbV represents a new family, proposed here as Abyssoviridae. MLeV represents a sister group to the other known coronaviruses. The importance of MLeV and AAbV for understanding nidovirus evolution, and the origin of terrestrial nidoviruses are discussed.
Keywords: Nidovirales; Protease; Protein expression; Proteinase; Readthrough; Transcriptome; Translation; Virus discovery.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures


References
-
- Bailey-Elkin B.A., Knaap R.C.M., Johnson G.G., Dalebout T.J., Ninaber D.K., Van Kasteren P.B., Bredenbeek P.J., Snijder E.J., Kikkert M., Mark B.L. Crystal structure of the middle east respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014;289:34667–34682. doi: 10.1074/jbc.M114.609644. - DOI - PMC - PubMed
-
- Brinton, M.A., Gulyaeva, A., Balasuriya, U.B.R., Dunowska, M., Faaberg, K.S., Goldberg, T., Leung, F.-C., Nauwynck, H.J., Snijder, E.J., Stadejek, T., Gorbalenya, A.E., 2017. ICTV Pending proposal 2017.012S. Expansion of the rank structure of the family Arteriviridae and renaming its taxa.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
