

Network-Based Approaches to Explore Complex Biological Systems towards Network Medicine
- PMID: 30200360
- PMCID: PMC6162385
- DOI: 10.3390/genes9090437
Network-Based Approaches to Explore Complex Biological Systems towards Network Medicine
Abstract
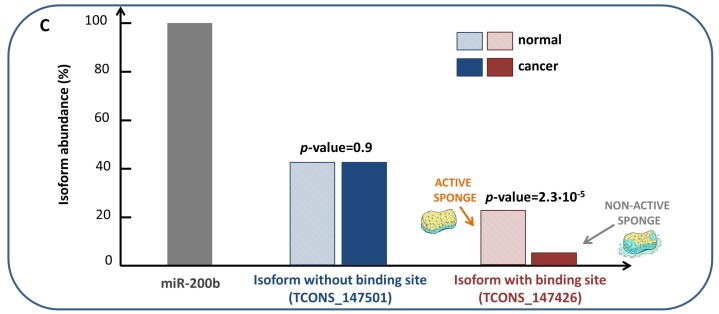
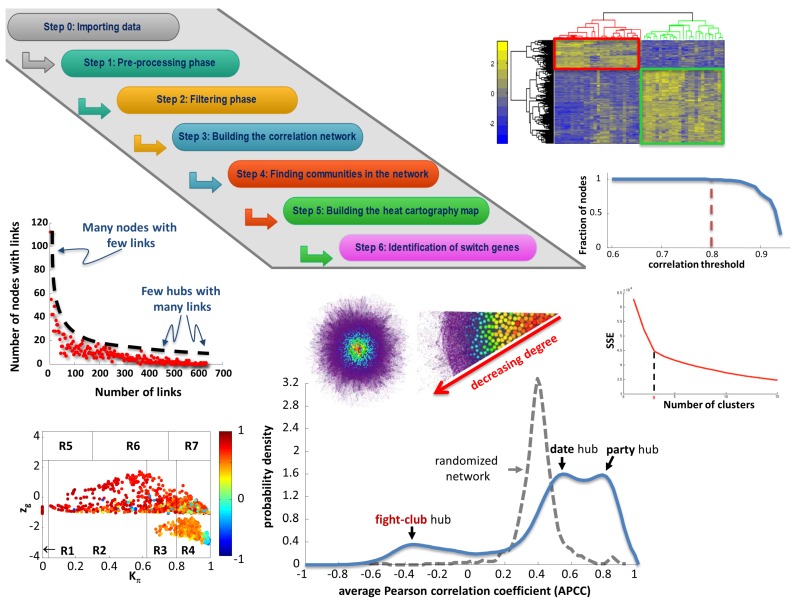
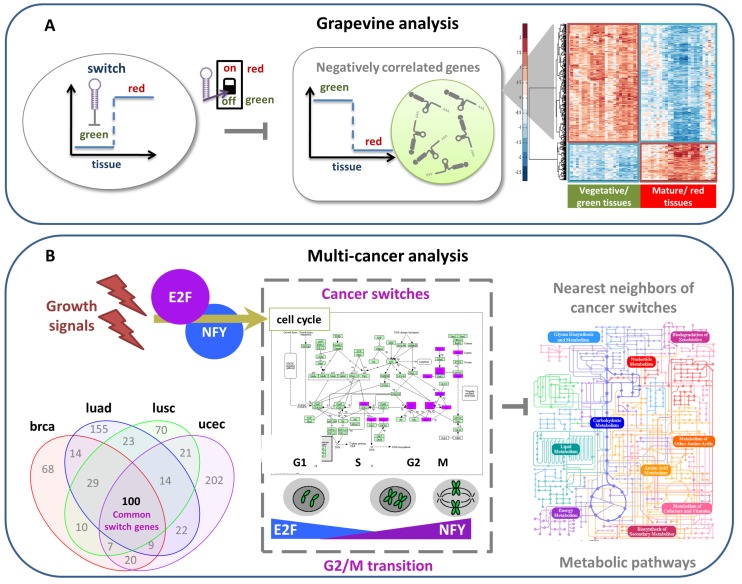
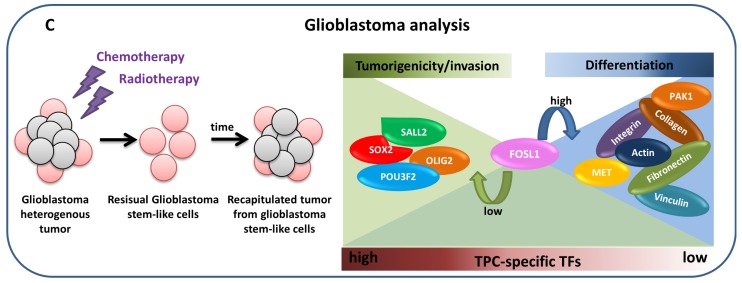
Network medicine relies on different types of networks: from the molecular level of protein⁻protein interactions to gene regulatory network and correlation studies of gene expression. Among network approaches based on the analysis of the topological properties of protein⁻protein interaction (PPI) networks, we discuss the widespread DIAMOnD (disease module detection) algorithm. Starting from the assumption that PPI networks can be viewed as maps where diseases can be identified with localized perturbation within a specific neighborhood (i.e., disease modules), DIAMOnD performs a systematic analysis of the human PPI network to uncover new disease-associated genes by exploiting the connectivity significance instead of connection density. The past few years have witnessed the increasing interest in understanding the molecular mechanism of post-transcriptional regulation with a special emphasis on non-coding RNAs since they are emerging as key regulators of many cellular processes in both physiological and pathological states. Recent findings show that coding genes are not the only targets that microRNAs interact with. In fact, there is a pool of different RNAs-including long non-coding RNAs (lncRNAs) -competing with each other to attract microRNAs for interactions, thus acting as competing endogenous RNAs (ceRNAs). The framework of regulatory networks provides a powerful tool to gather new insights into ceRNA regulatory mechanisms. Here, we describe a data-driven model recently developed to explore the lncRNA-associated ceRNA activity in breast invasive carcinoma. On the other hand, a very promising example of the co-expression network is the one implemented by the software SWIM (switch miner), which combines topological properties of correlation networks with gene expression data in order to identify a small pool of genes-called switch genes-critically associated with drastic changes in cell phenotype. Here, we describe SWIM tool along with its applications to cancer research and compare its predictions with DIAMOnD disease genes.
Keywords: PPI network; bioinformatics; ceRNA; gene co-expression network; network medicine; regulatory network.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
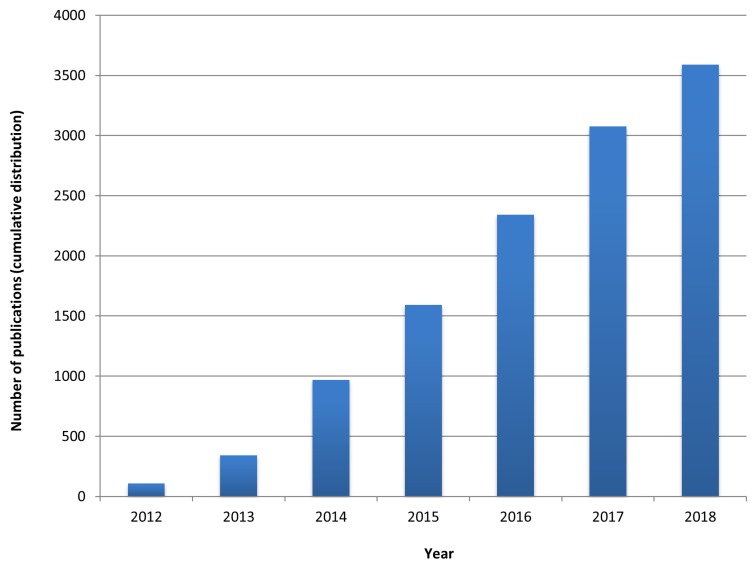
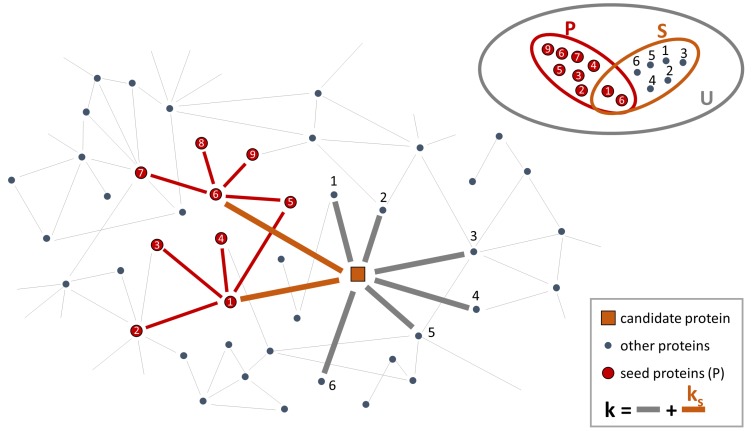
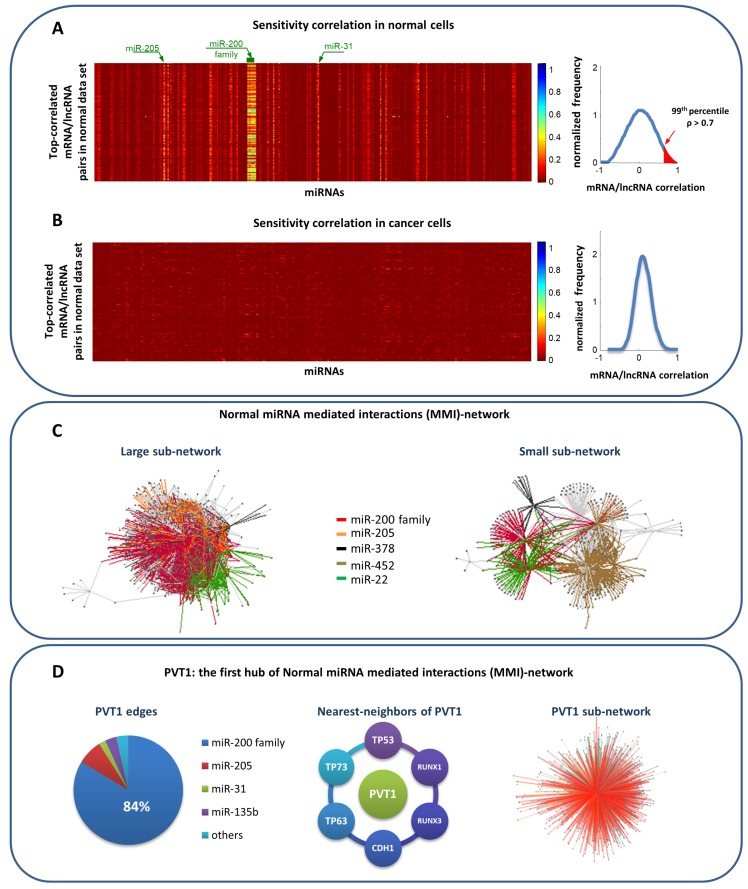
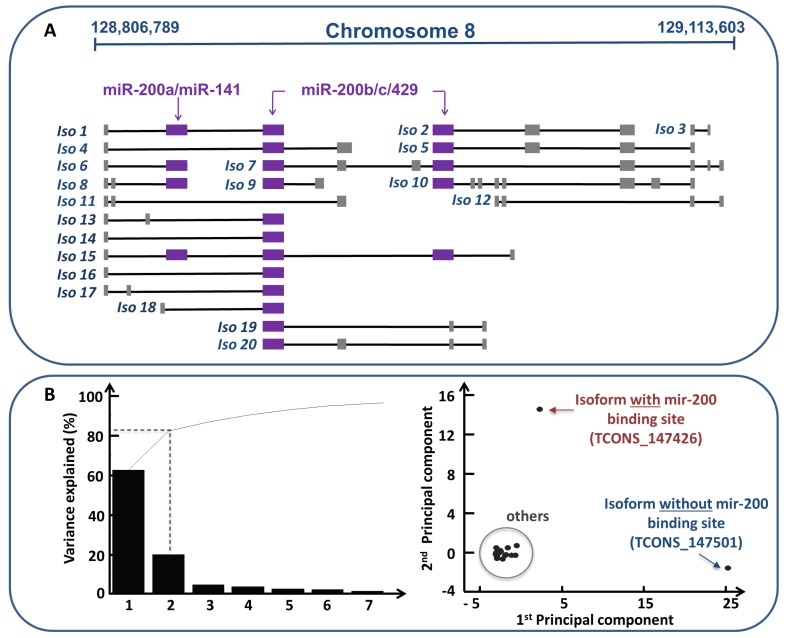
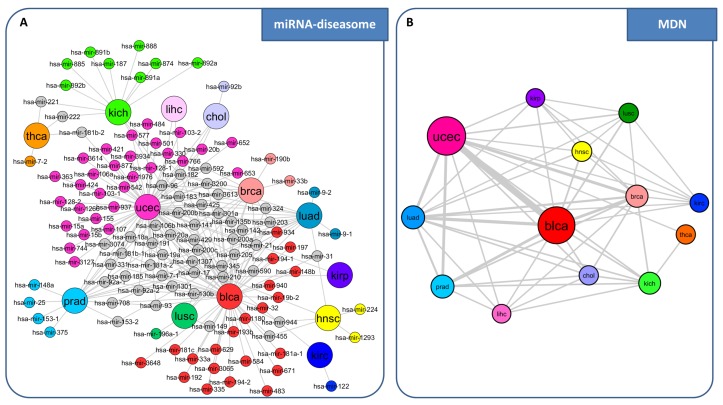
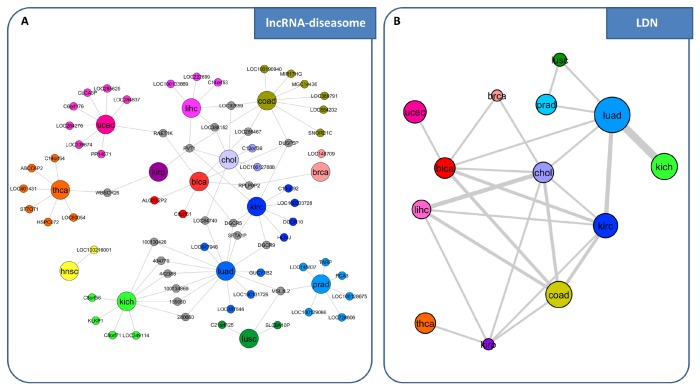
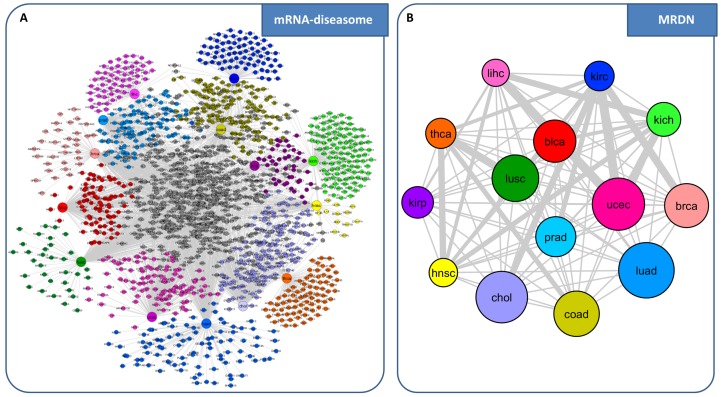
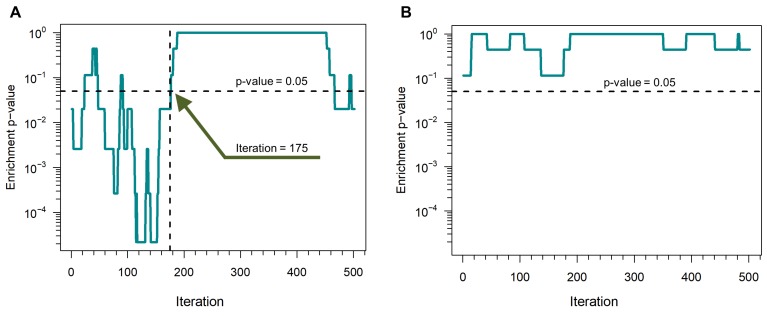
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
