

Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA
- PMID: 30201725
- PMCID: PMC6166824
- DOI: 10.1073/pnas.1806447115
Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA
Abstract
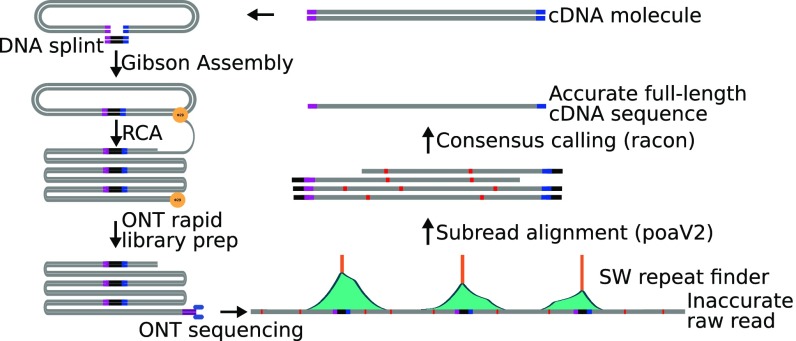
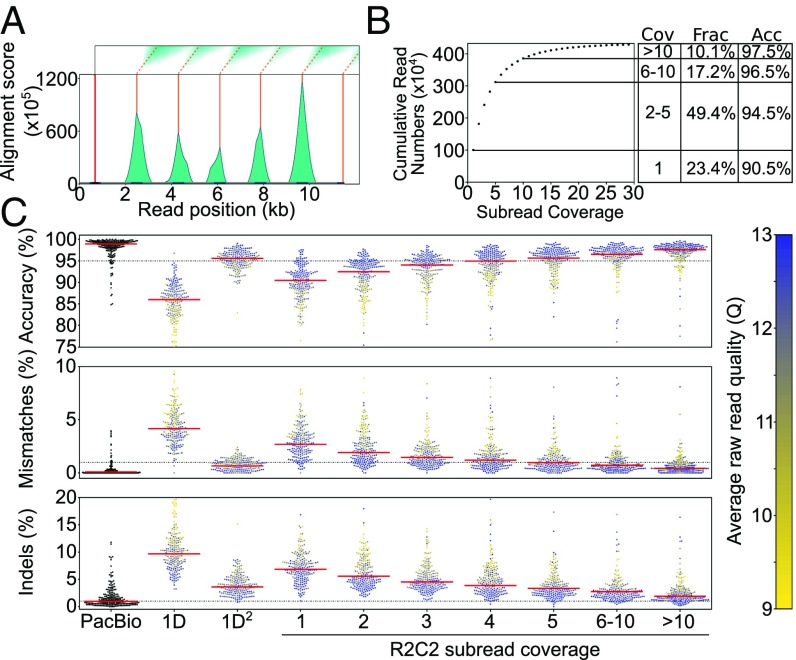
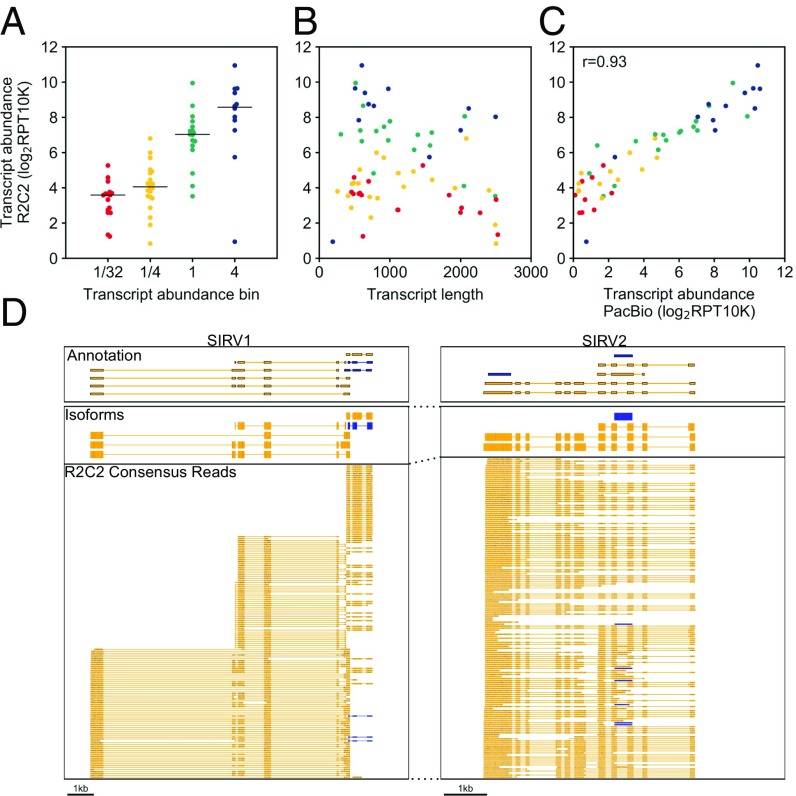
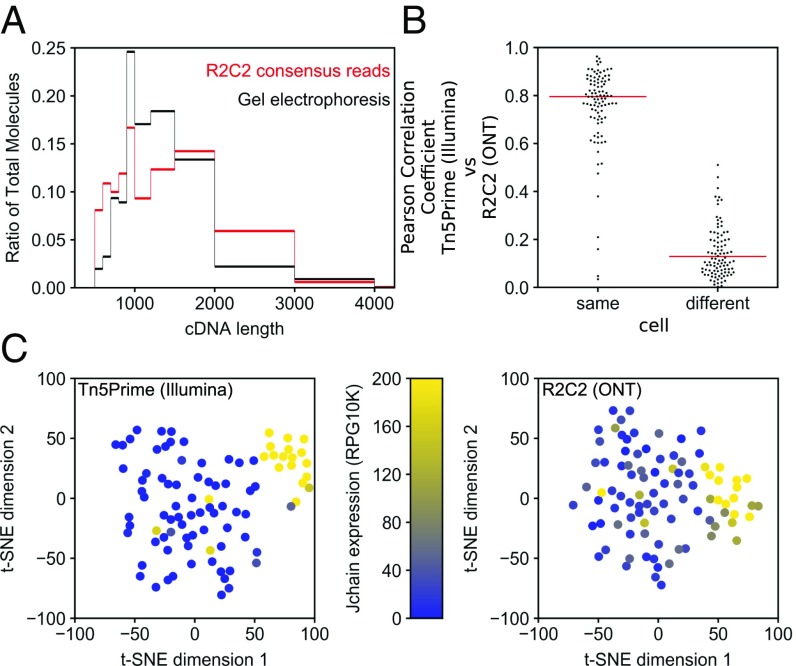
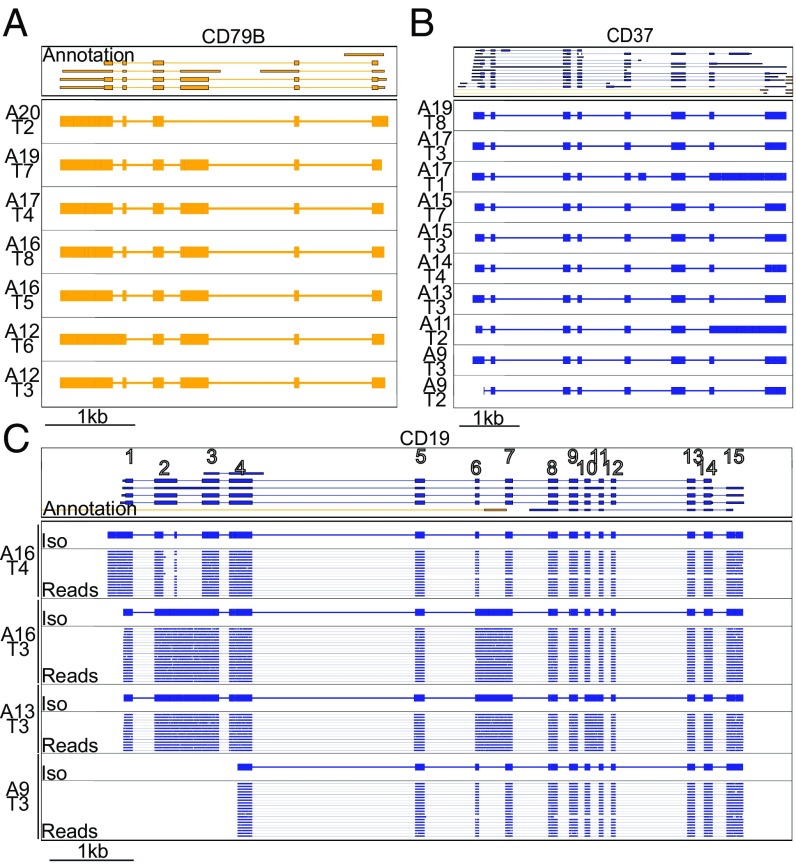
High-throughput short-read sequencing has revolutionized how transcriptomes are quantified and annotated. However, while Illumina short-read sequencers can be used to analyze entire transcriptomes down to the level of individual splicing events with great accuracy, they fall short of analyzing how these individual events are combined into complete RNA transcript isoforms. Because of this shortfall, long-distance information is required to complement short-read sequencing to analyze transcriptomes on the level of full-length RNA transcript isoforms. While long-read sequencing technology can provide this long-distance information, there are issues with both Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) long-read sequencing technologies that prevent their widespread adoption. Briefly, PacBio sequencers produce low numbers of reads with high accuracy, while ONT sequencers produce higher numbers of reads with lower accuracy. Here, we introduce and validate a long-read ONT-based sequencing method. At the same cost, our Rolling Circle Amplification to Concatemeric Consensus (R2C2) method generates more accurate reads of full-length RNA transcript isoforms than any other available long-read sequencing method. These reads can then be used to generate isoform-level transcriptomes for both genome annotation and differential expression analysis in bulk or single-cell samples.
Keywords: B cells; full-length cDNA sequencing; isoforms; nanopore sequencing; single-cell transcriptomics.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
Conflict of interest statement: C.V., R.E.G., T.P., and R.V. have filed a provisional patent on the methodology described in the paper. The other authors have nothing to declare.
Figures
Comment in
-
Improving long-read accuracy.Nat Methods. 2018 Nov;15(11):860. doi: 10.1038/s41592-018-0204-y. Nat Methods. 2018. PMID: 30377358 No abstract available.
References
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 2008;5:621–628. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
