

Terminal exon characterization with TECtool reveals an abundance of cell-specific isoforms
- PMID: 30202060
- PMCID: PMC7611301
- DOI: 10.1038/s41592-018-0114-z
Terminal exon characterization with TECtool reveals an abundance of cell-specific isoforms
Abstract
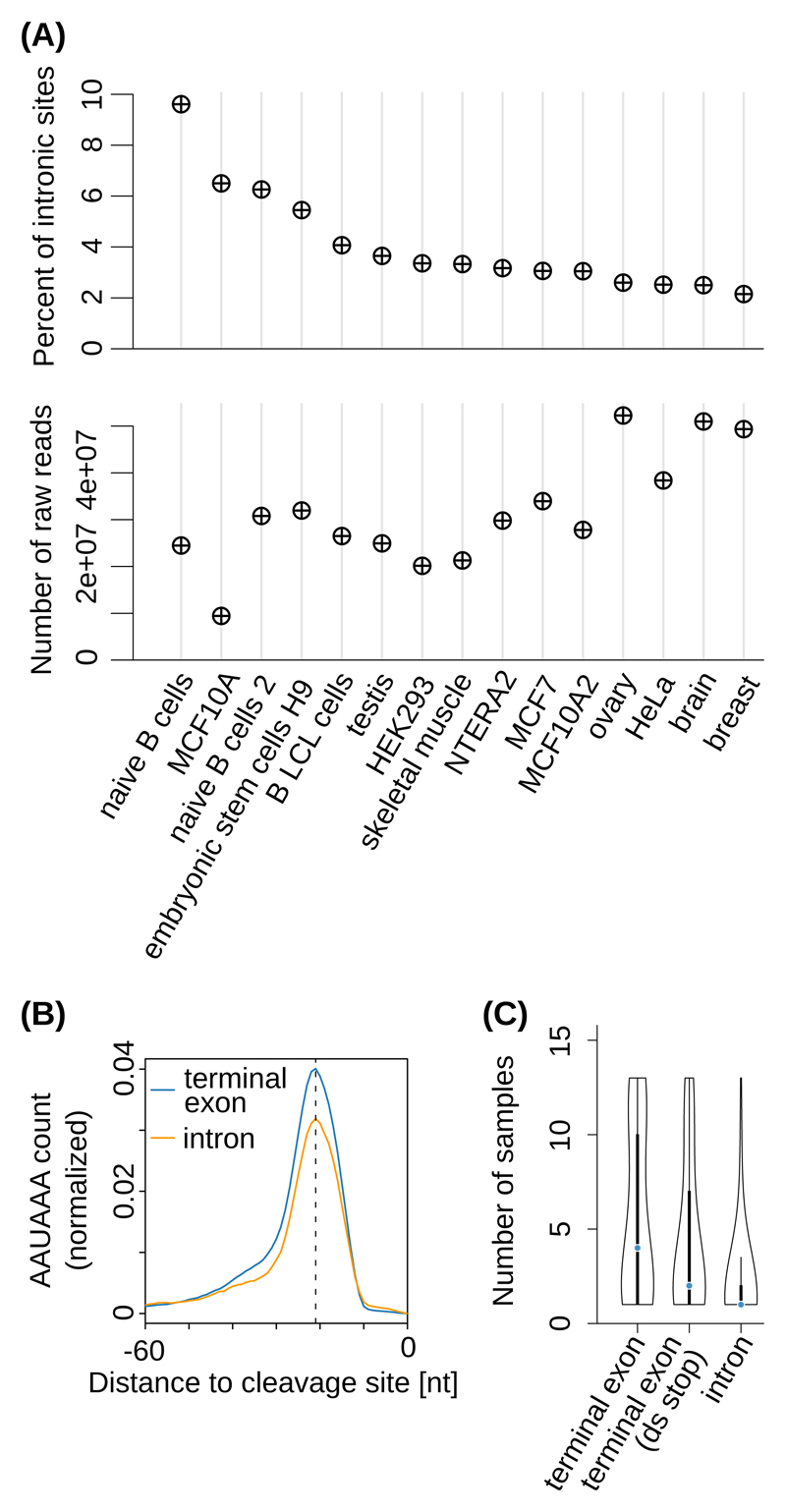
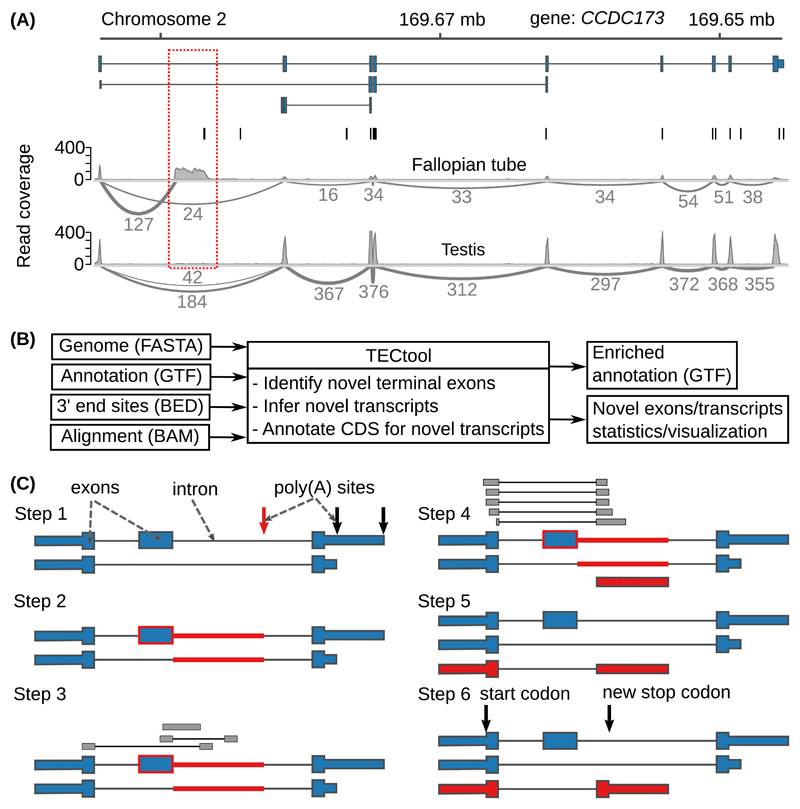
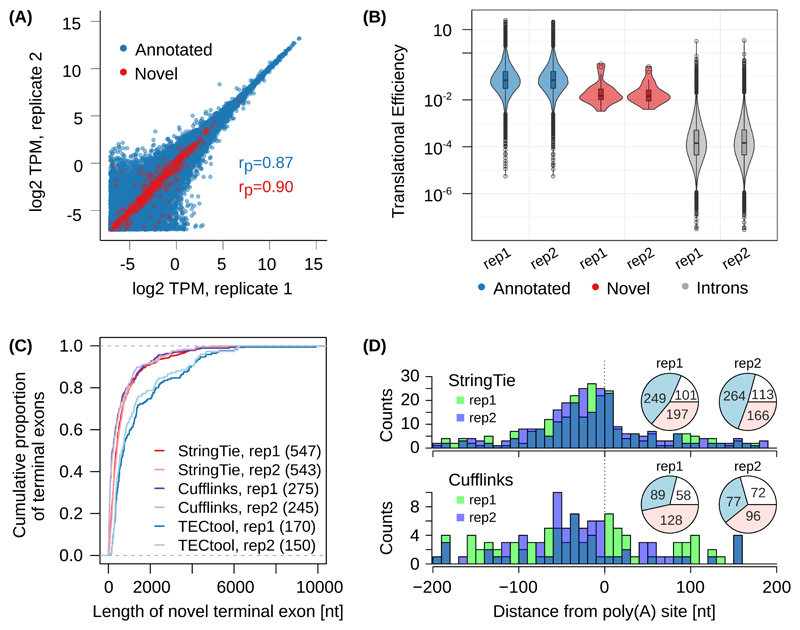
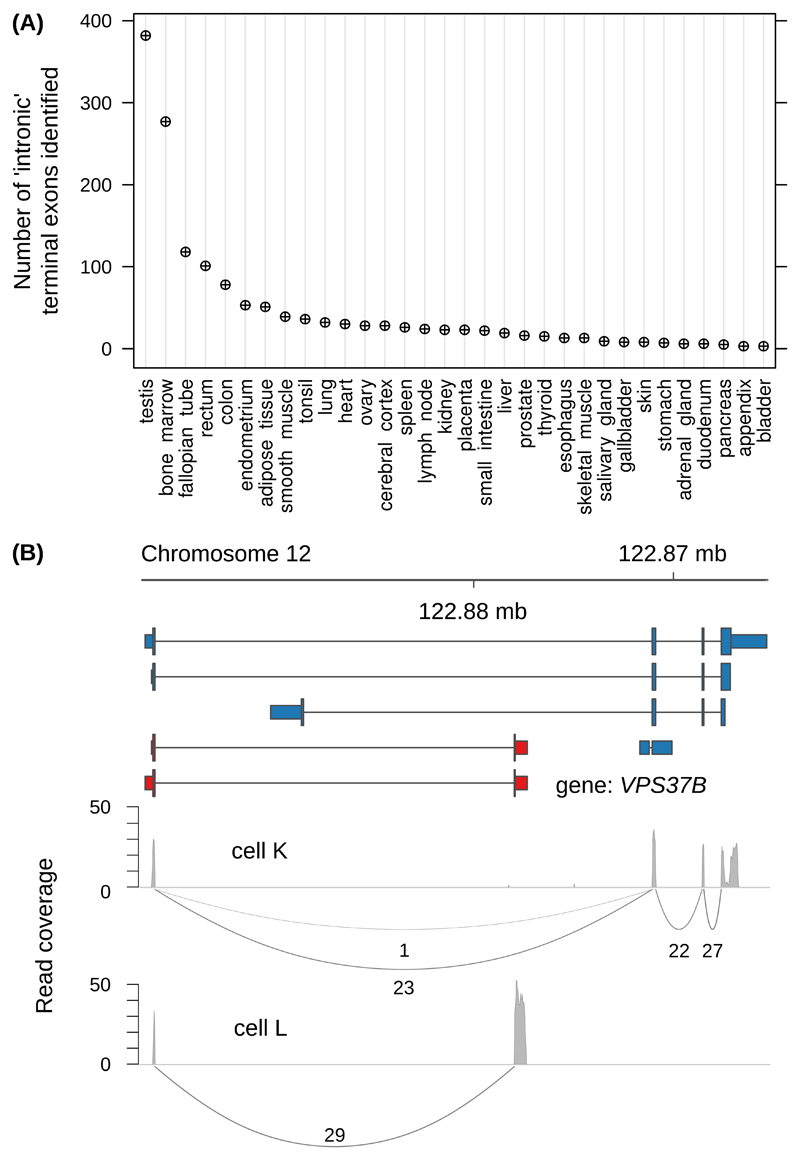
Sequencing of RNA 3' ends has uncovered numerous sites that do not correspond to the termination sites of known transcripts. Through their 3' untranslated regions, protein-coding RNAs interact with RNA-binding proteins and microRNAs, which regulate many properties, including RNA stability and subcellular localization. We developed the terminal exon characterization (TEC) tool ( http://tectool.unibas.ch ), which can be used with RNA-sequencing data from any species for which a genome annotation that includes sites of RNA cleavage and polyadenylation is available. We discovered hundreds of previously unknown isoforms and cell-type-specific terminal exons in human cells. Ribosome profiling data revealed that many of these isoforms were translated. By applying TECtool to single-cell sequencing data, we found that the newly identified isoforms were expressed in subpopulations of cells. Thus, TECtool enables the identification of previously unknown isoforms in well-studied cell systems and in rare cell types.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Hausser J, Zavolan M. Identification and consequences of miRNA--target interactions—beyond repression of gene expression. Nat Rev Genet. 2014;15:599. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
