

Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD
- PMID: 30202070
- PMCID: PMC6131187
- DOI: 10.1038/s41467-018-06115-2
Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD
Abstract
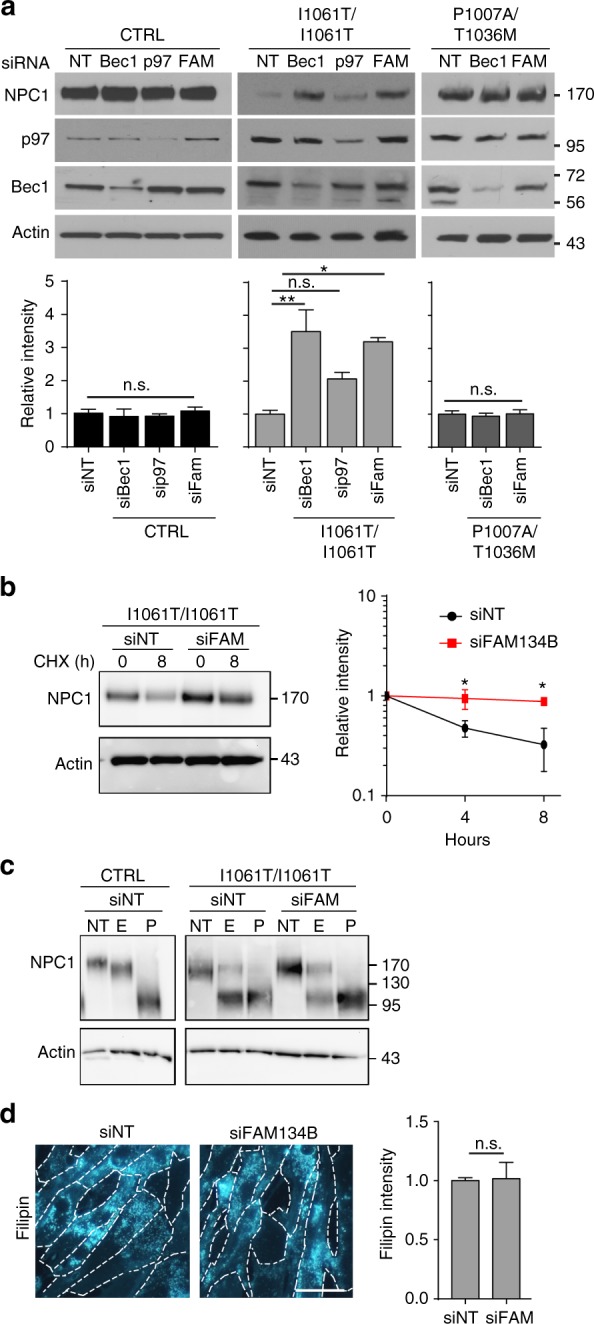
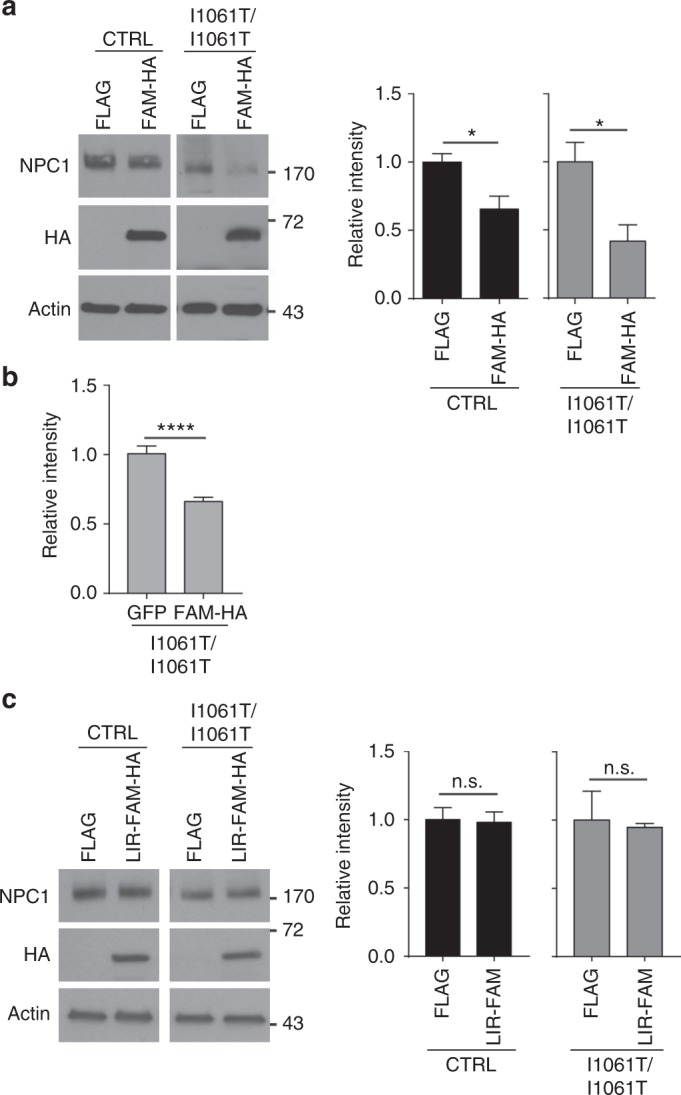
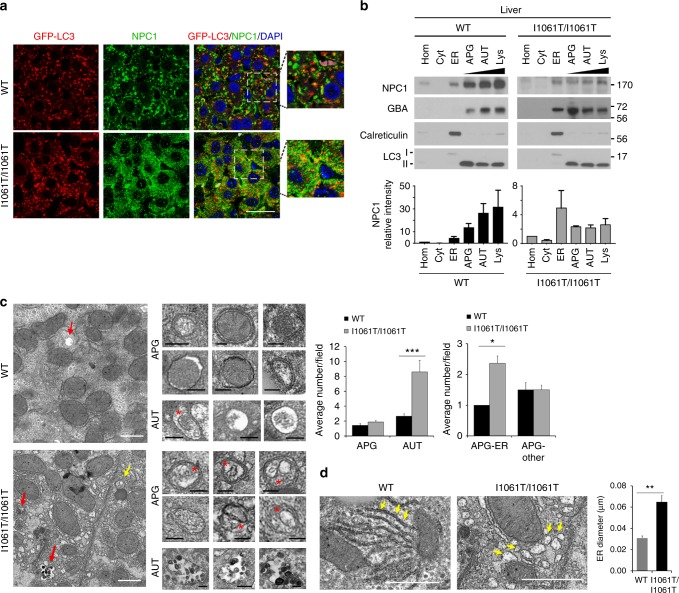
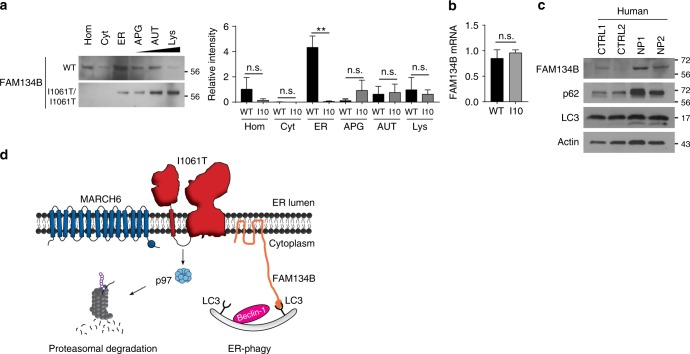
Niemann-Pick type C disease is a fatal, progressive neurodegenerative disorder caused by loss-of-function mutations in NPC1, a multipass transmembrane glycoprotein essential for intracellular lipid trafficking. We sought to define the cellular machinery controlling degradation of the most common disease-causing mutant, I1061T NPC1. We show that this mutant is degraded, in part, by the proteasome following MARCH6-dependent ERAD. Unexpectedly, we demonstrate that I1061T NPC1 is also degraded by a recently described autophagic pathway called selective ER autophagy (ER-phagy). We establish the importance of ER-phagy both in vitro and in vivo, and identify I1061T as a misfolded endogenous substrate for this FAM134B-dependent process. Subcellular fractionation of I1061T Npc1 mouse tissues and analysis of human samples show alterations of key components of ER-phagy, including FAM134B. Our data establish that I1061T NPC1 is recognized in the ER and degraded by two different pathways that function in a complementary fashion to regulate protein turnover.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
