

Experimental and computational framework for a dynamic protein atlas of human cell division
- PMID: 30202089
- PMCID: PMC6556381
- DOI: 10.1038/s41586-018-0518-z
Experimental and computational framework for a dynamic protein atlas of human cell division
Abstract
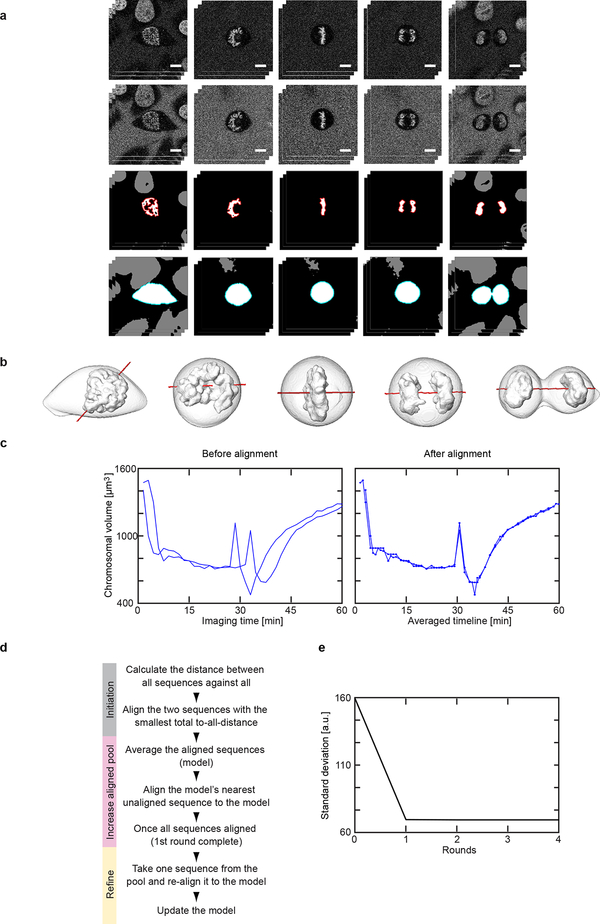
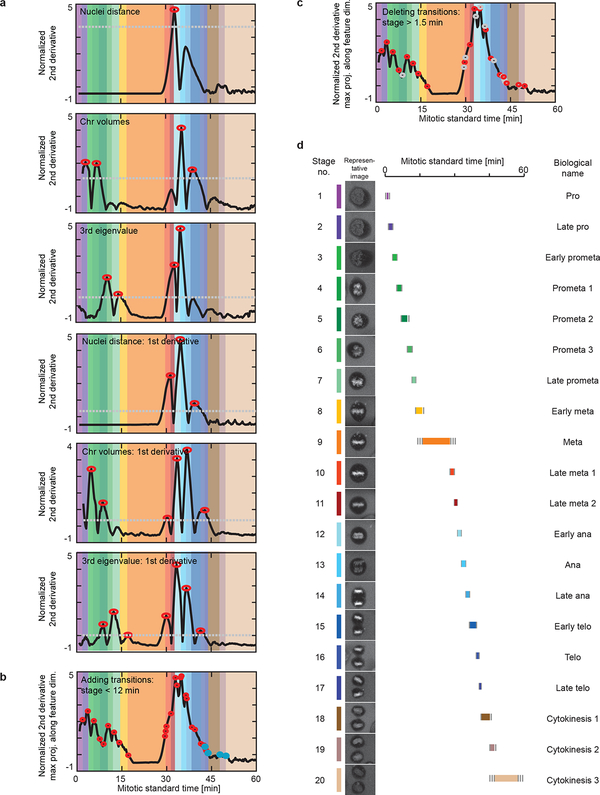
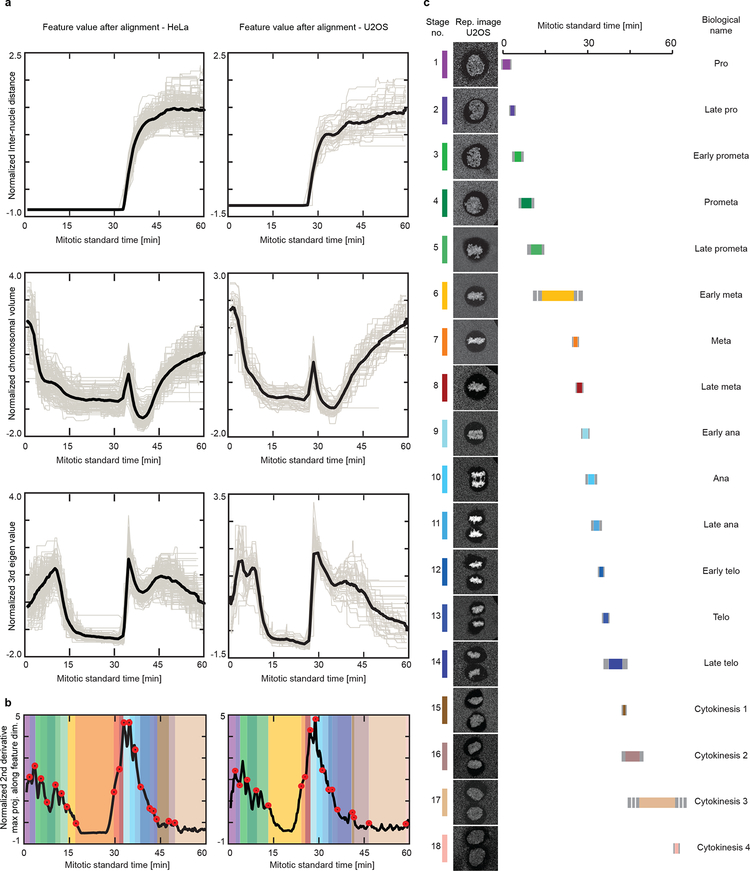
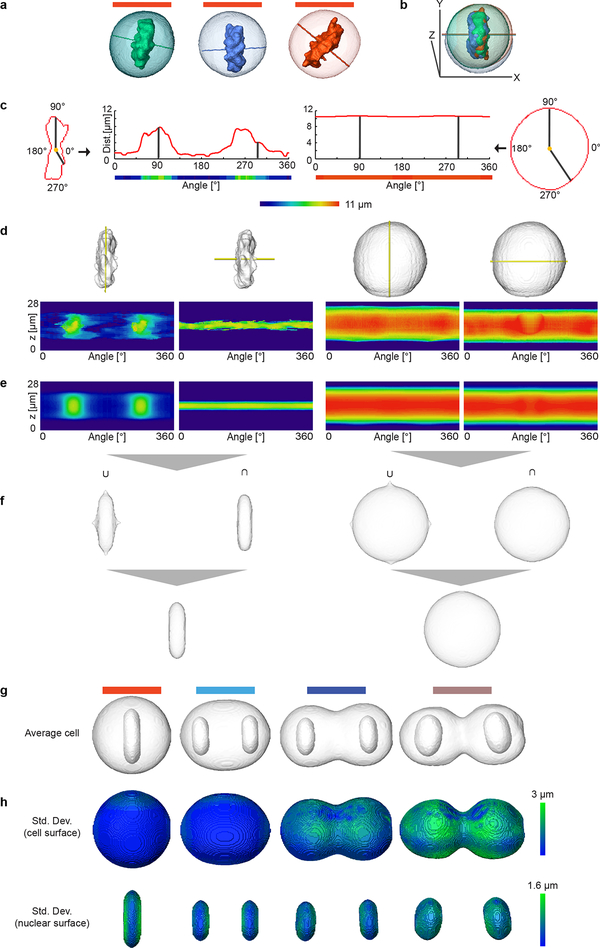
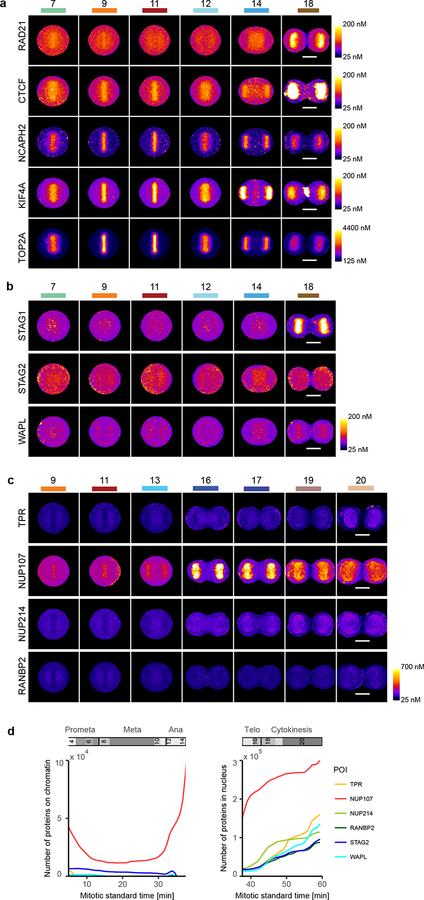
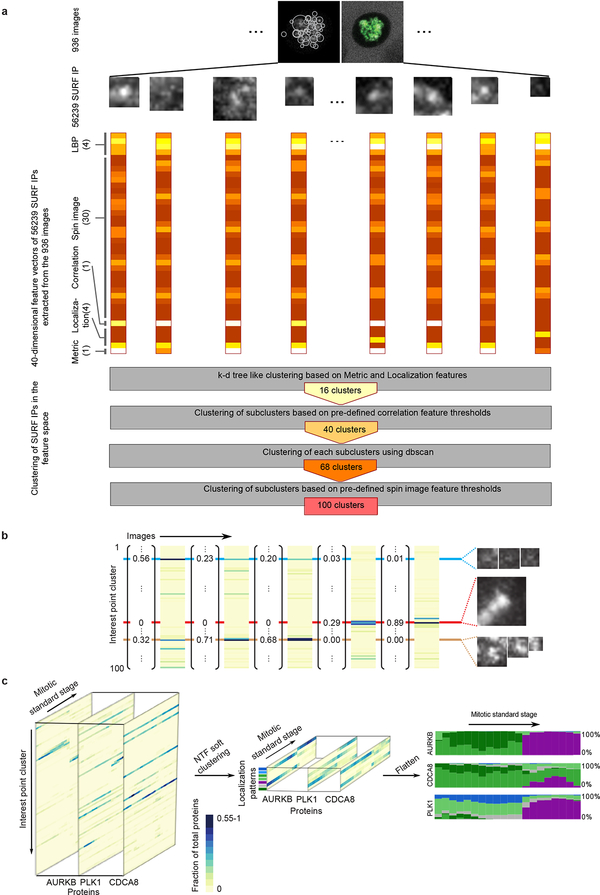
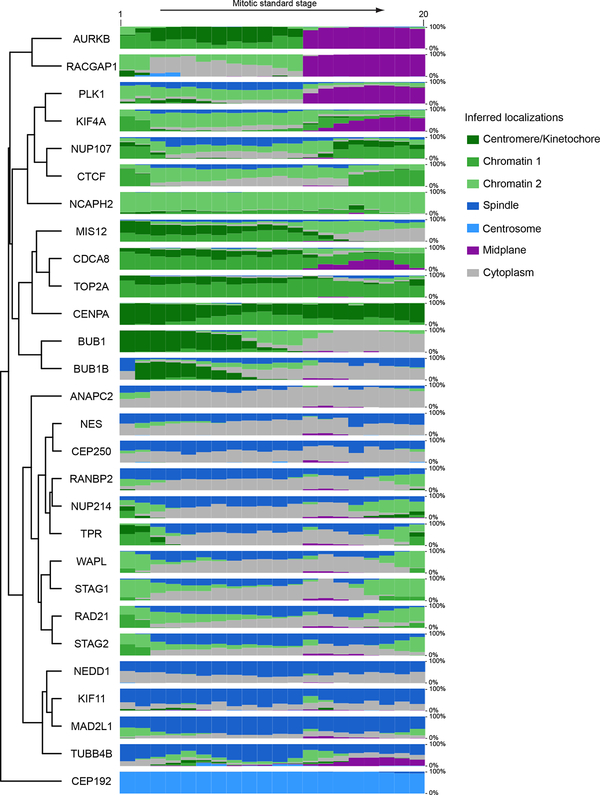
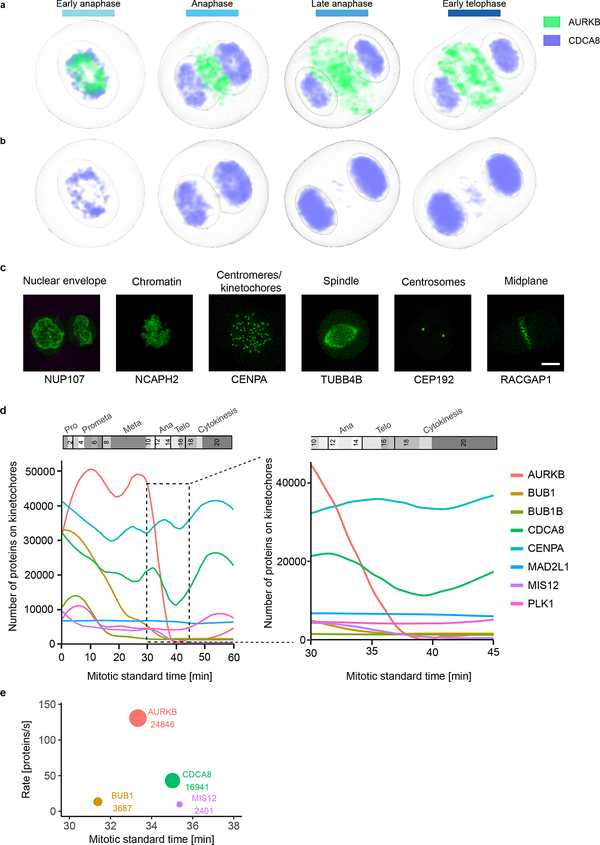
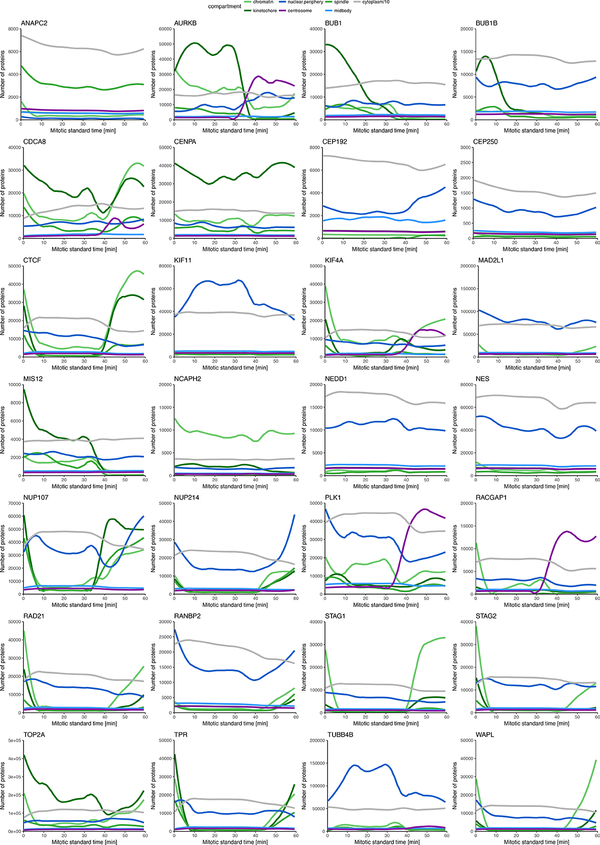
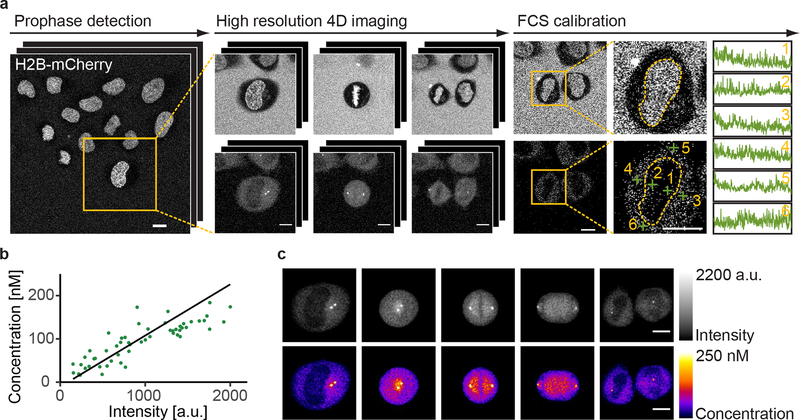
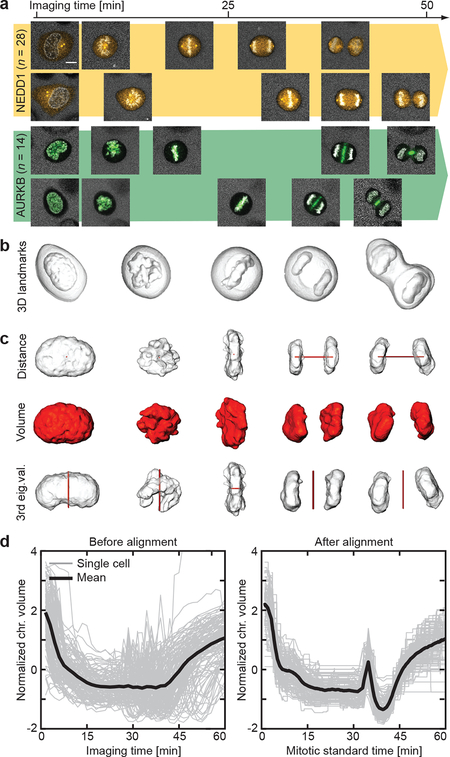
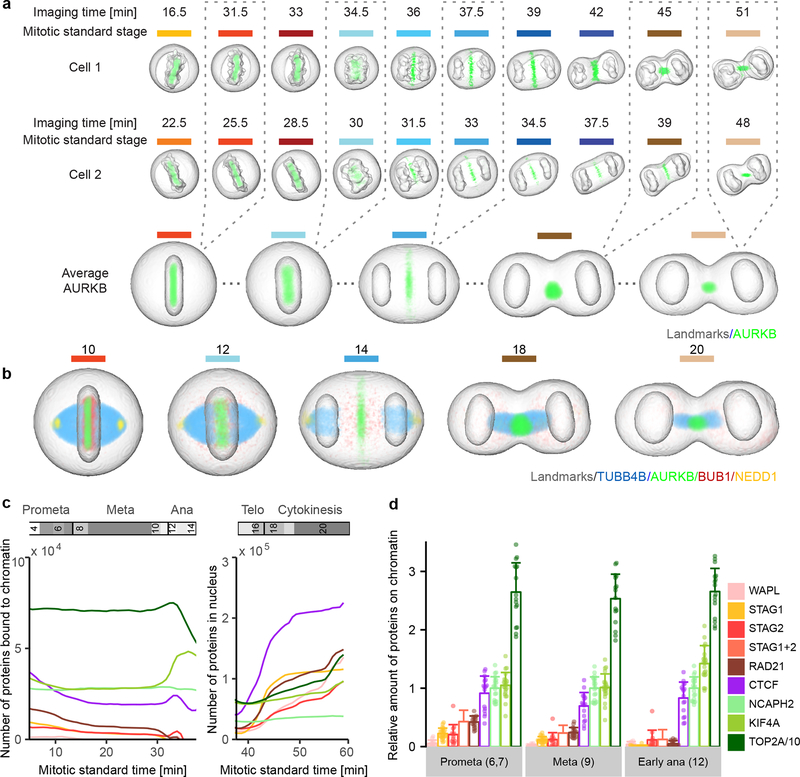
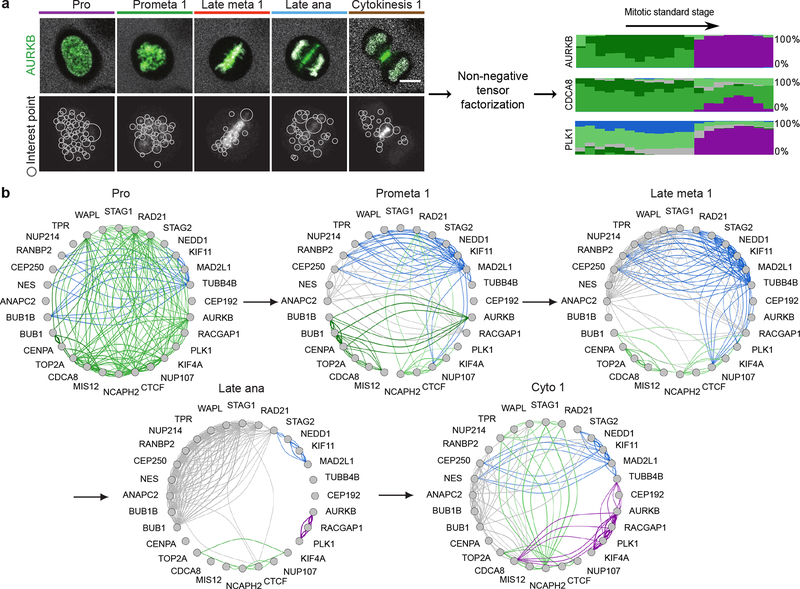
Essential biological functions, such as mitosis, require tight coordination of hundreds of proteins in space and time. Localization, the timing of interactions and changes in cellular structure are all crucial to ensure the correct assembly, function and regulation of protein complexes1-4. Imaging of live cells can reveal protein distributions and dynamics but experimental and theoretical challenges have prevented the collection of quantitative data, which are necessary for the formulation of a model of mitosis that comprehensively integrates information and enables the analysis of the dynamic interactions between the molecular parts of the mitotic machinery within changing cellular boundaries. Here we generate a canonical model of the morphological changes during the mitotic progression of human cells on the basis of four-dimensional image data. We use this model to integrate dynamic three-dimensional concentration data of many fluorescently knocked-in mitotic proteins, imaged by fluorescence correlation spectroscopy-calibrated microscopy5. The approach taken here to generate a dynamic protein atlas of human cell division is generic; it can be applied to systematically map and mine dynamic protein localization networks that drive cell division in different cell types, and can be conceptually transferred to other cellular functions.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
