

CLINICAL CHARACTERIZATION OF STARGARDT DISEASE PATIENTS WITH THE p.N1868I ABCA4 MUTATION
- PMID: 30204727
- PMCID: PMC6548695
- DOI: 10.1097/IAE.0000000000002316
CLINICAL CHARACTERIZATION OF STARGARDT DISEASE PATIENTS WITH THE p.N1868I ABCA4 MUTATION
Abstract
Purpose: To investigate the Stargardt disease phenotype associated with an unusually common and "extremely hypomorphic" ABCA4 variant, p.N1868I.
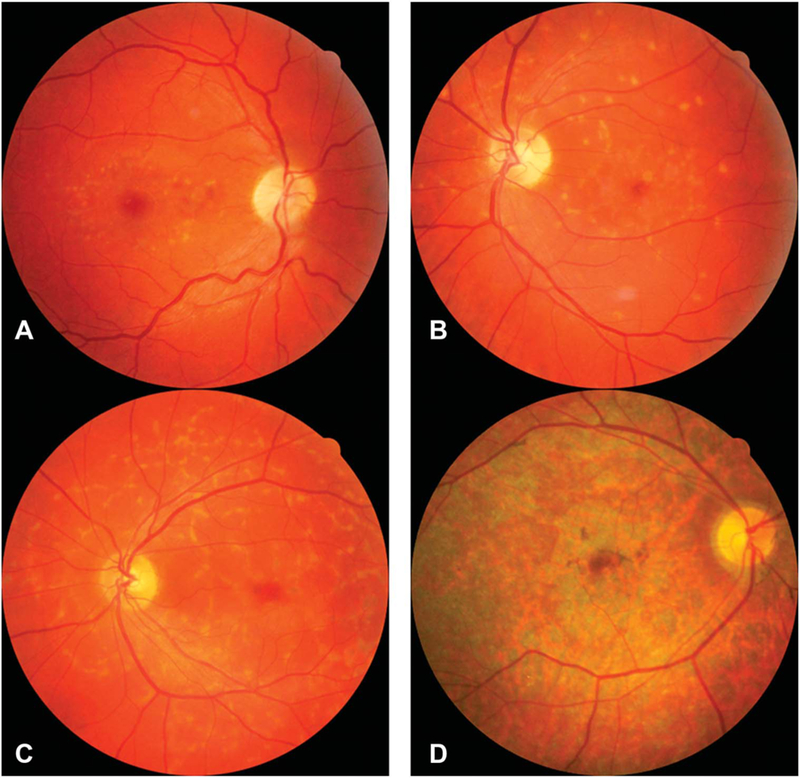
Methods: The charts of 27 patients with p.N1868I on one allele and a severe/deleterious mutation on the other allele were reviewed. Subjective age of onset, best-corrected visual acuity, and stage of disease were recorded for all 27 patients, 18 of whom had multiple visits. When available, fundus photography, spectral domain optical coherence tomography, fundus autofluorescence, full-field electroretinograms, Goldmann visual fields, and fluorescein angiography were included. Five families with multiple affected members were analyzed.
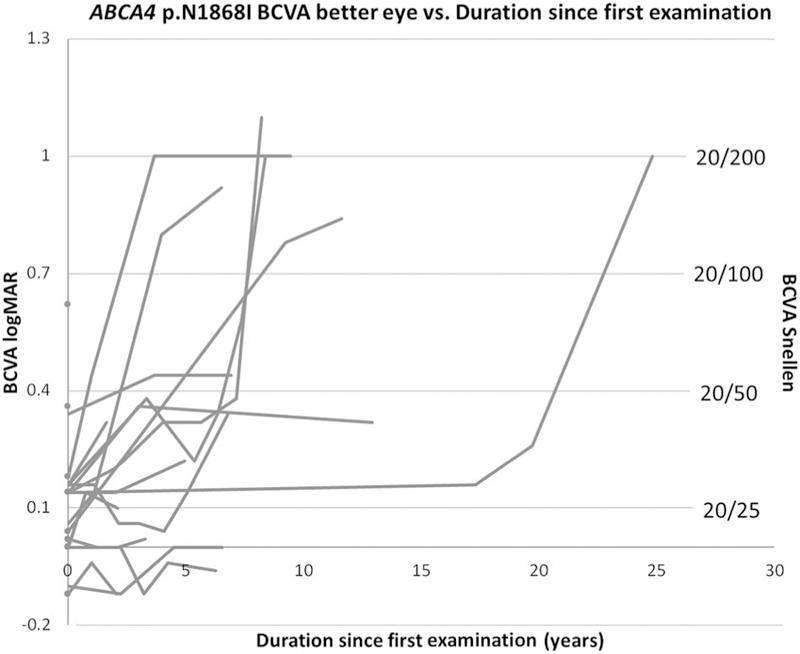
Results: The median age at symptom onset was 41.5 years, and 3 p.N1868I patients had not developed visual symptoms as of the most recent eye examination. Median best-corrected visual acuity in the better-seeing eye at baseline was 20/25, and the median duration from symptom onset to legal blindness was 25 years. The five families described in this study demonstrated clinically significant intrafamilial variability, and affected family members who did not share the p.N1868I variant had relatively more severe phenotypes.
Conclusion: This study demonstrates the consistency of foveal sparing, the variation in age at onset, the intrafamilial variability, and the prognosis with regard to visual acuity in p.N1868I-associated Stargardt disease.
Conflict of interest statement
None of the authors has any financial/conflicting interests to disclose.
Figures





References
-
- Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997;15: 236–246. - PubMed
-
- Fishman GA, Farber M, Patel BS, Derlacki DJ. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology 1987;94:809–814. - PubMed
-
- Noble KG, Carr RE. Stargardt’s disease and fundus flavimaculatus. Arch Ophthalmol 1979;97:1281–1285. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
