

The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers
- PMID: 30205045
- PMCID: PMC6327853
- DOI: 10.1016/j.ccell.2018.08.008
The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers
Abstract
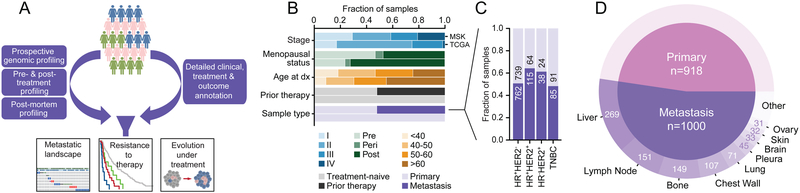
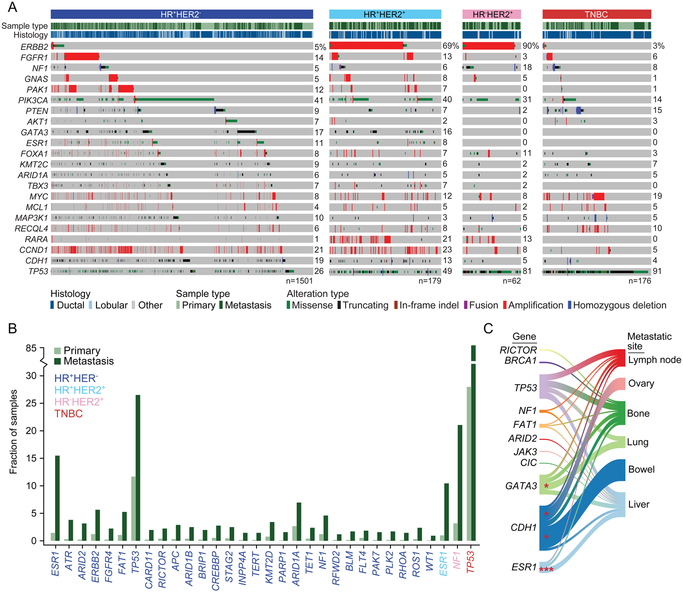
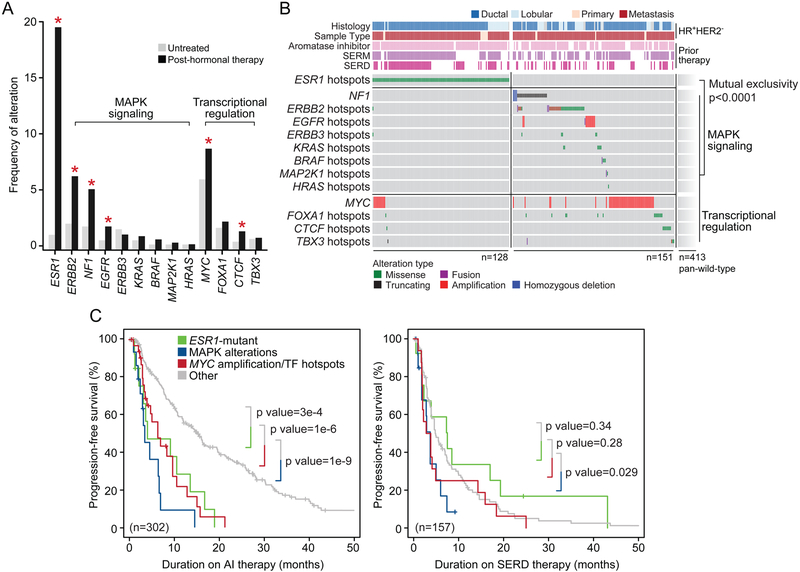
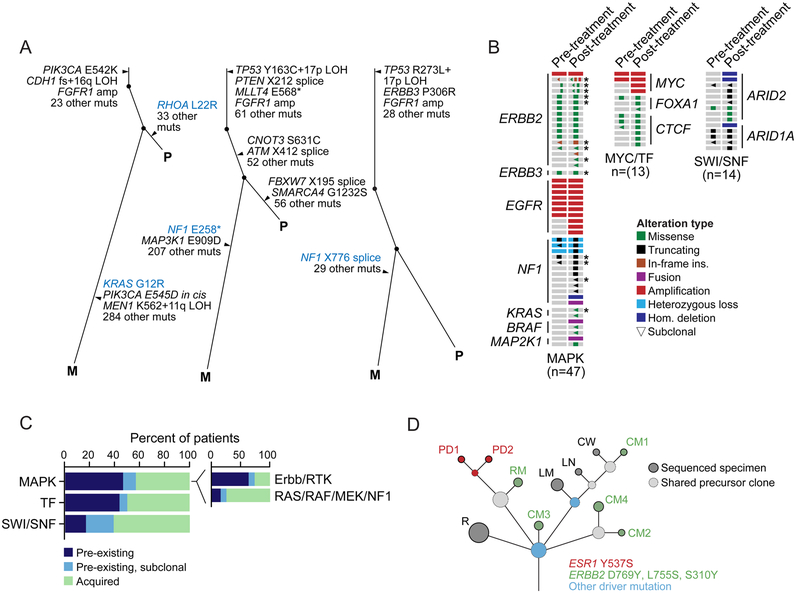
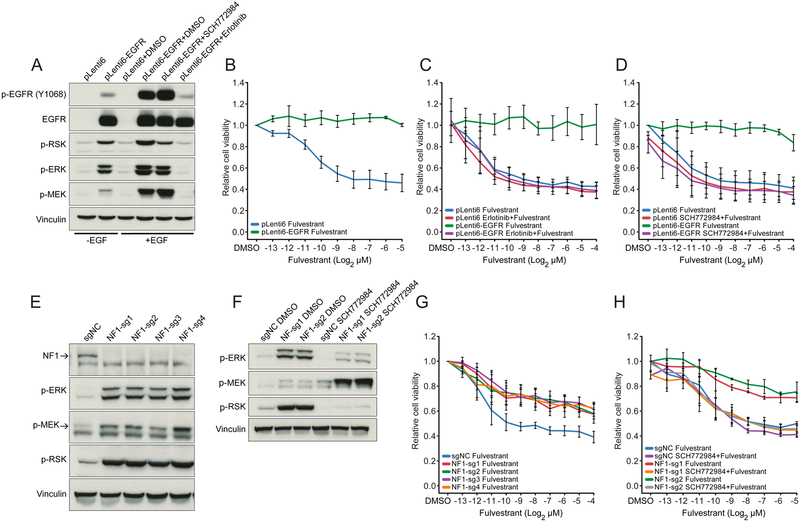
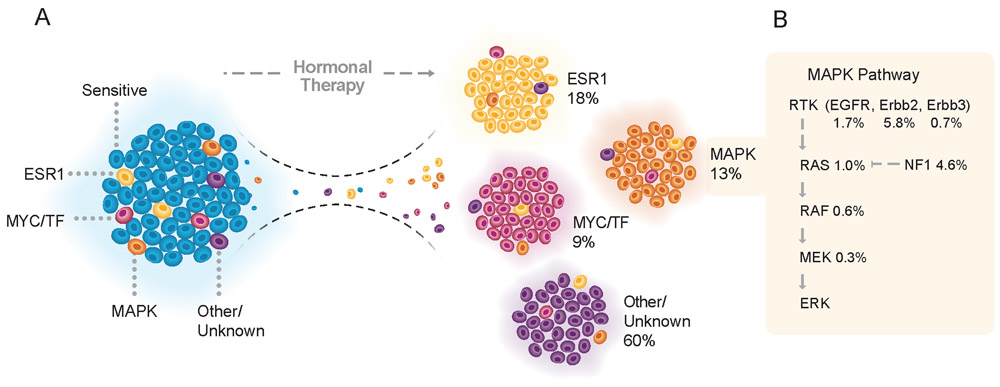
We integrated the genomic sequencing of 1,918 breast cancers, including 1,501 hormone receptor-positive tumors, with detailed clinical information and treatment outcomes. In 692 tumors previously exposed to hormonal therapy, we identified an increased number of alterations in genes involved in the mitogen-activated protein kinase (MAPK) pathway and in the estrogen receptor transcriptional machinery. Activating ERBB2 mutations and NF1 loss-of-function mutations were more than twice as common in endocrine resistant tumors. Alterations in other MAPK pathway genes (EGFR, KRAS, among others) and estrogen receptor transcriptional regulators (MYC, CTCF, FOXA1, and TBX3) were also enriched. Altogether, these alterations were present in 22% of tumors, mutually exclusive with ESR1 mutations, and associated with a shorter duration of response to subsequent hormonal therapies.
Keywords: breast cancer; cancer genomics; endocrine resistance; integrative genomics analysis; metastasis.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
-
- Andre F, Campone M, Ciruelos EM, Iwata H, Liobl S, Rugo HS, Wilke C, Mills D, Chol M, Longin A, et al. (2016). SOLAR-1: A phase III study of alpelisib + fulvestrant in men and postmenopausal women with HR+/HER2− advanced breast cancer (BC) progressing on or after prior aromatase inhibitor therapy. J Clin Oncol 34, TPS618–TPS618.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
