

Induction of HIF-1α by HIV-1 Infection in CD4+ T Cells Promotes Viral Replication and Drives Extracellular Vesicle-Mediated Inflammation
- PMID: 30206166
- PMCID: PMC6134101
- DOI: 10.1128/mBio.00757-18
Induction of HIF-1α by HIV-1 Infection in CD4+ T Cells Promotes Viral Replication and Drives Extracellular Vesicle-Mediated Inflammation
Abstract
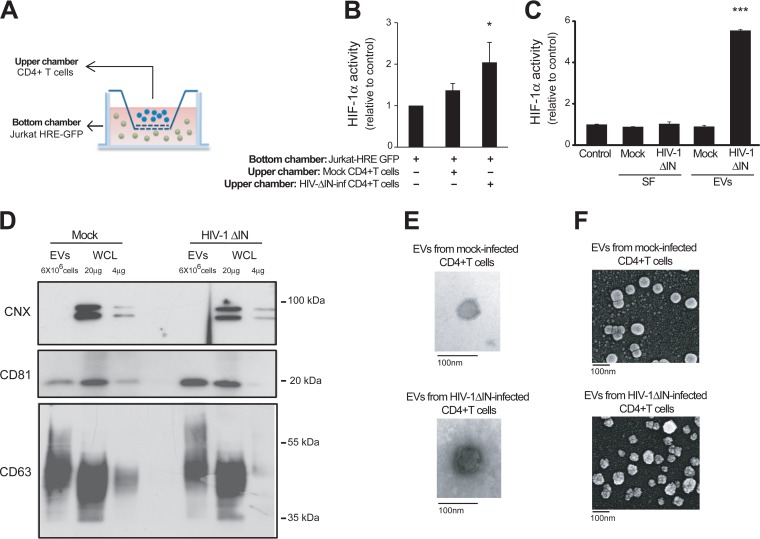
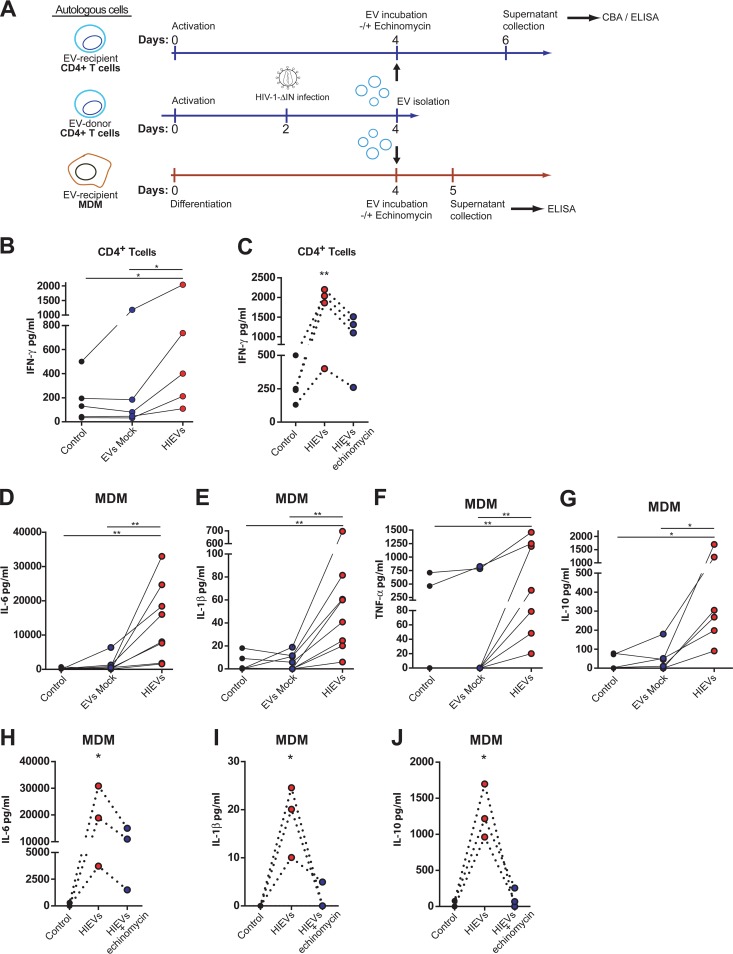
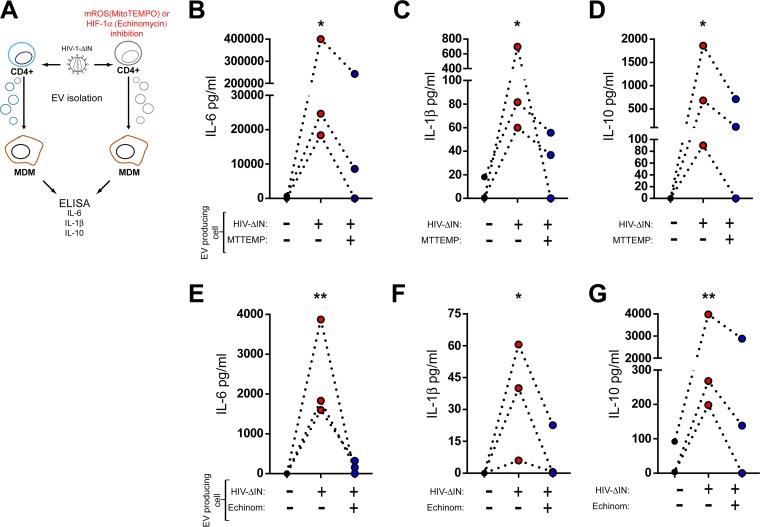
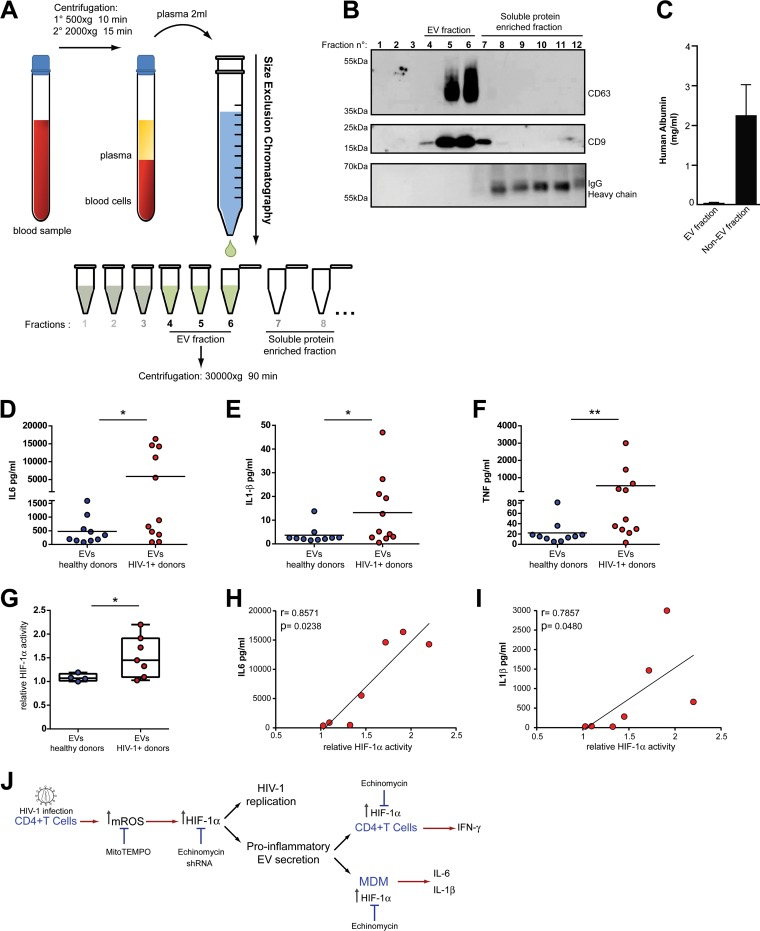
Chronic immune activation and inflammation are hallmarks of HIV-1 infection and a major cause of serious non-AIDS events in HIV-1-infected individuals on antiretroviral treatment (ART). Herein, we show that cytosolic double-stranded DNA (dsDNA) generated in infected CD4+ T cells during the HIV-1 replication cycle promotes the mitochondrial reactive oxygen species (ROS)-dependent stabilization of the transcription factor hypoxia-inducible factor 1α (HIF-1α), which in turn, enhances viral replication. Furthermore, we show that induction of HIF-1α promotes the release of extracellular vesicles (EVs). These EVs foster inflammation by inducing the secretion of gamma interferon by bystander CD4+ T cells and secretion of interleukin 6 (IL-6) and IL-1β by bystander macrophages through an HIF-1α-dependent pathway. Remarkably, EVs obtained from plasma samples from HIV-1-infected individuals also induced HIF-1α activity and inflammation. Overall, this study demonstrates that HIF-1α plays a crucial role in HIV-1 pathogenesis by promoting viral replication and the release of EVs that orchestrate lymphocyte- and macrophage-mediated inflammatory responses.IMPORTANCE Human immunodeficiency virus type 1 (HIV-1) is a very important global pathogen that preferentially targets CD4+ T cells and causes acquired immunodeficiency syndrome (AIDS) if left untreated. Although antiretroviral treatment efficiently suppresses viremia, markers of immune activation and inflammation remain higher in HIV-1-infected patients than in uninfected individuals. The hypoxia-inducible factor 1α (HIF-1α) is a transcription factor that plays a fundamental role in coordinating cellular metabolism and function. Here we show that HIV-1 infection induces HIF-1α activity and that this transcription factor upholds HIV-1 replication. Moreover, we demonstrate that HIF-1α plays a key role in HIV-1-associated inflammation by promoting the release of extracellular vesicles which, in turn, trigger the secretion of inflammatory mediators by noninfected bystander lymphocytes and macrophages. In summary, we identify that the coordinated actions of HIF-1α and extracellular vesicles promote viral replication and inflammation, thus contributing to HIV-1 pathogenesis.
Keywords: CD4+ T lymphocyte; extracellular vesicles; human immunodeficiency virus; hypoxia-inducible factor 1 alpha; inflammation; macrophage.
Copyright © 2018 Duette et al.
Figures
References
-
- Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA. 1998. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280:427–431. doi: 10.1126/science.280.5362.427. - DOI - PubMed
-
- Deeks SG, Gange SJ, Kitahata MM, Saag MS, Justice AC, Hogg RS, Eron JJ, Brooks JT, Rourke SB, Gill MJ, Bosch RJ, Benson CA, Collier AC, Martin JN, Klein MB, Jacobson LP, Rodriguez B, Sterling TR, Kirk GD, Napravnik S, Rachlis AR, Calzavara LM, Horberg MA, Silverberg MJ, Gebo KA, Kushel MB, Goedert JJ, McKaig RG, Moore RD. 2009. Trends in multidrug treatment failure and subsequent mortality among antiretroviral therapy-experienced patients with HIV infection in North America. Clin Infect Dis 49:1582–1590. doi: 10.1086/644768. - DOI - PMC - PubMed
-
- Duffau P, Wittkop L, Lazaro E, le Marec F, Cognet C, Blanco P, Moreau JF, Dauchy FA, Cazanave C, Vandenhende MA, Bonnet F, Thiebaut R, Pellegrin I, ANRS CO3 Aquitaine Cohort Study Group . 2015. Association of immune-activation and senescence markers with non-AIDS-defining comorbidities in HIV-suppressed patients. AIDS 29:2099–2108. doi: 10.1097/QAD.0000000000000807. - DOI - PubMed
-
- Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, Ledergerber B, Lundgren J, Neuhaus J, Nixon D, Paton NI, Neaton JD, INSIGHT SMART Study Group . 2008. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 5:e203. doi: 10.1371/journal.pmed.0050203. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
