

Anti-TNFR1 targeting in humanized mice ameliorates disease in a model of multiple sclerosis
- PMID: 30206422
- PMCID: PMC6133964
- DOI: 10.1038/s41598-018-31957-7
Anti-TNFR1 targeting in humanized mice ameliorates disease in a model of multiple sclerosis
Abstract
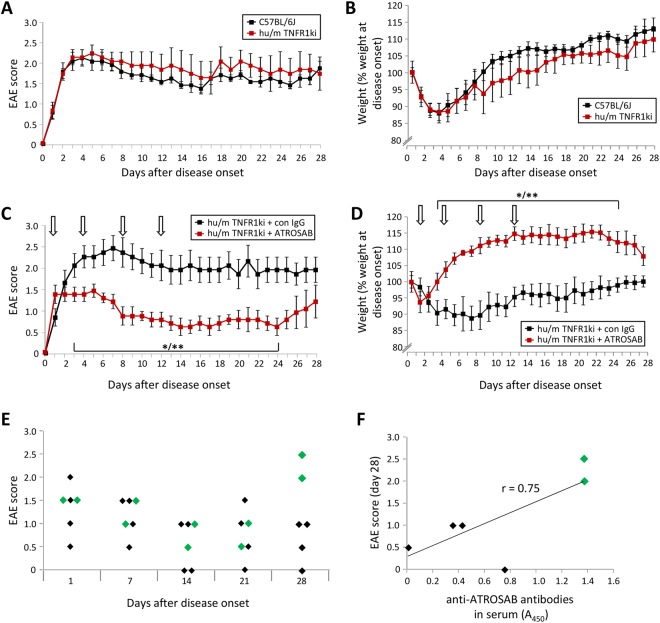
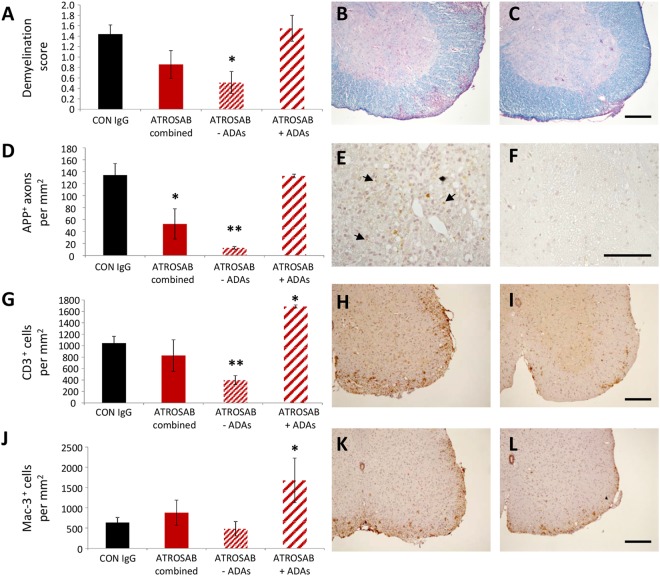
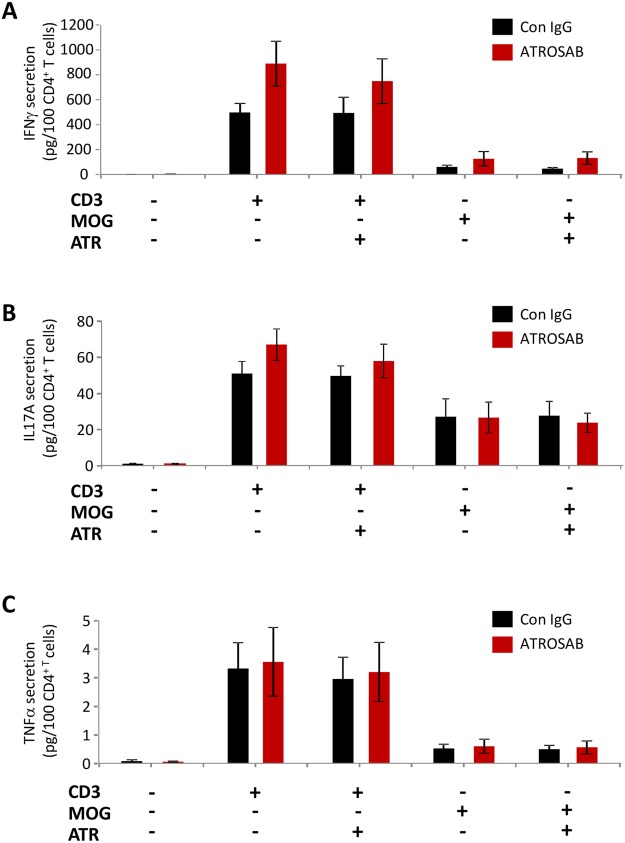
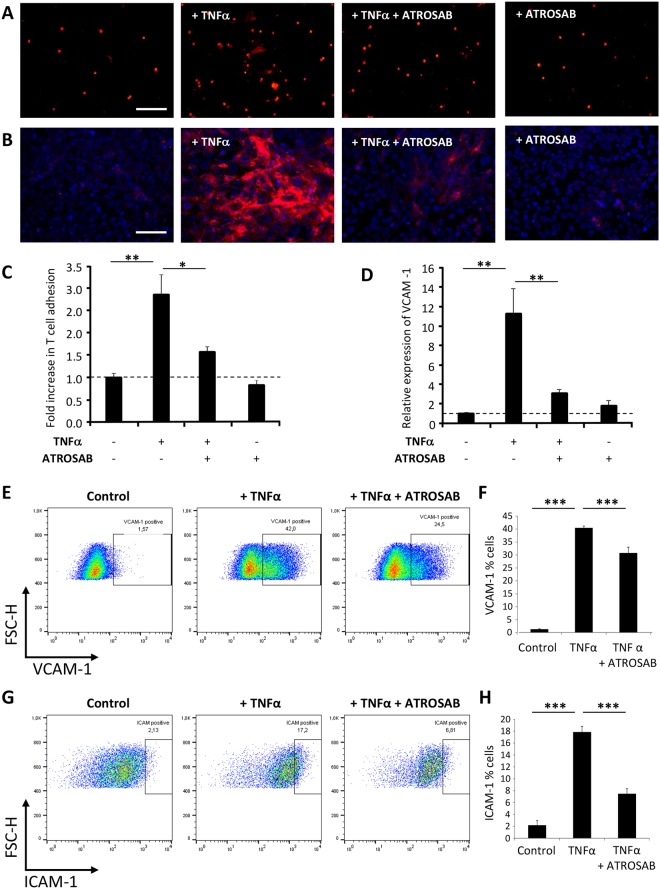
Tumour necrosis factor (TNF) signalling is mediated via two receptors, TNF-receptor 1 (TNFR1) and TNF-receptor 2 (TNFR2), which work antithetically to balance CNS immune responses involved in autoimmune diseases such as multiple sclerosis. To determine the therapeutic potential of selectively inhibiting TNFR1 in mice with experimental autoimmune encephalomyelitis, we used chimeric human/mouse TNFR1 knock-in mice allowing the evaluation of antagonistic anti-human TNFR1 antibody efficacy. Treatment of mice after onset of disease with ATROSAB resulted in a robust amelioration of disease severity, correlating with reduced central nervous system immune cell infiltration. Long-term efficacy of treatment was achieved by treatment with the parental mouse anti-human TNFR1 antibody, H398, and extended by subsequent re-treatment of mice following relapse. Our data support the hypothesis that anti-TNFR1 therapy restricts immune cell infiltration across the blood-brain barrier through the down-regulation of TNF-induced adhesion molecules, rather than altering immune cell composition or activity. Collectively, we demonstrate the potential for anti-human TNFR1 therapies to effectively modulate immune responses in autoimmune disease.
Conflict of interest statement
R.K., A.H. and K. Pfizenmaier are named inventors of patents covering the ATROSAB technology. A.H. is owner of Baliopharm, which is developing ATROSAB for clinical use. The authors have no additional financial interests. S.K.W., R. Fairless, O.M., P.C.L., K. Pichi, R. Fischer, U.L.M.E., B.W., N.R., P-O.C. and R.D. declare no potential conflict of interest.
Figures



Similar articles
-
Co-modulation of TNFR1 and TNFR2 in an animal model of multiple sclerosis.J Neuroinflammation. 2023 Apr 30;20(1):100. doi: 10.1186/s12974-023-02784-z. J Neuroinflammation. 2023. PMID: 37122019 Free PMC article.
-
The TNFR1 Antagonist Atrosimab Is Therapeutic in Mouse Models of Acute and Chronic Inflammation.Front Immunol. 2021 Jul 7;12:705485. doi: 10.3389/fimmu.2021.705485. eCollection 2021. Front Immunol. 2021. PMID: 34305946 Free PMC article.
-
Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis.PLoS One. 2014 Feb 28;9(2):e90117. doi: 10.1371/journal.pone.0090117. eCollection 2014. PLoS One. 2014. PMID: 24587232 Free PMC article.
-
Distinct modes of TNF signaling through its two receptors in health and disease.J Leukoc Biol. 2020 Jun;107(6):893-905. doi: 10.1002/JLB.2MR0120-510R. Epub 2020 Feb 21. J Leukoc Biol. 2020. PMID: 32083339 Review.
-
Selective inhibition of Tumor necrosis factor receptor-1 (TNFR1) for the treatment of autoimmune diseases.Cytokine Growth Factor Rev. 2020 Oct;55:80-85. doi: 10.1016/j.cytogfr.2020.03.002. Epub 2020 Apr 12. Cytokine Growth Factor Rev. 2020. PMID: 32327345 Review.
Cited by
-
Immunosuppressant Therapies in COVID-19: Is the TNF Axis an Alternative?Pharmaceuticals (Basel). 2022 May 17;15(5):616. doi: 10.3390/ph15050616. Pharmaceuticals (Basel). 2022. PMID: 35631442 Free PMC article. Review.
-
Targeting Differential Roles of Tumor Necrosis Factor Receptors as a Therapeutic Strategy for Glaucoma.Front Immunol. 2022 May 16;13:857812. doi: 10.3389/fimmu.2022.857812. eCollection 2022. Front Immunol. 2022. PMID: 35651608 Free PMC article. Review.
-
Anti-inflammatory treatment in MPN: targeting TNFR1 and TNFR2 in JAK2-V617F-induced disease.Blood Adv. 2021 Dec 14;5(23):5349-5359. doi: 10.1182/bloodadvances.2021004438. Blood Adv. 2021. PMID: 34592754 Free PMC article.
-
Apoptosis-Inducing TNF Superfamily Ligands for Cancer Therapy.Cancers (Basel). 2021 Mar 27;13(7):1543. doi: 10.3390/cancers13071543. Cancers (Basel). 2021. PMID: 33801589 Free PMC article. Review.
-
Insights into the biology and therapeutic implications of TNF and regulatory T cells.Nat Rev Rheumatol. 2021 Aug;17(8):487-504. doi: 10.1038/s41584-021-00639-6. Epub 2021 Jul 5. Nat Rev Rheumatol. 2021. PMID: 34226727 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
