

Robust activation of microhomology-mediated end joining for precision gene editing applications
- PMID: 30208061
- PMCID: PMC6152997
- DOI: 10.1371/journal.pgen.1007652
Robust activation of microhomology-mediated end joining for precision gene editing applications
Abstract
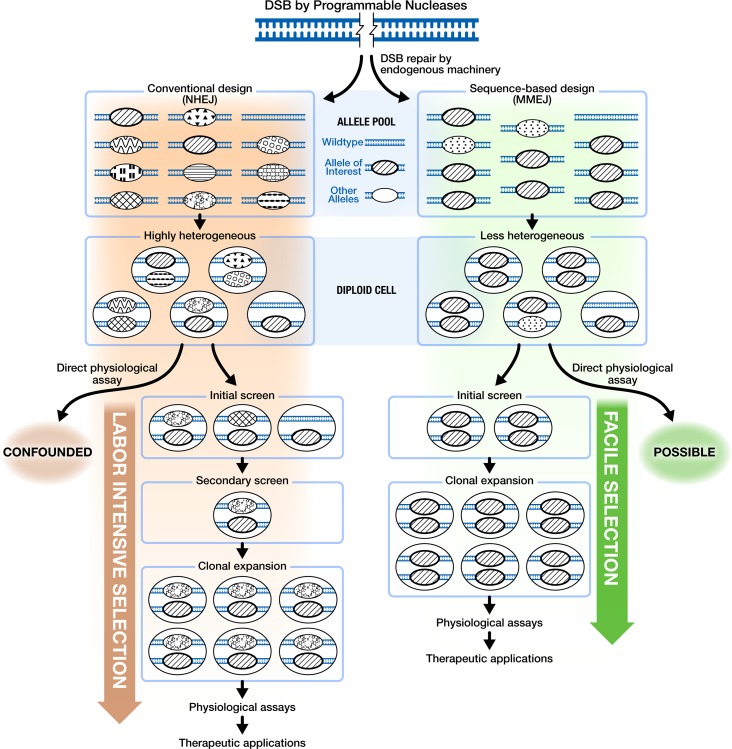
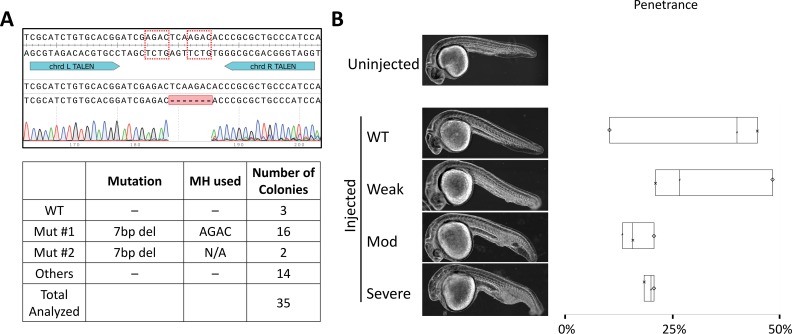
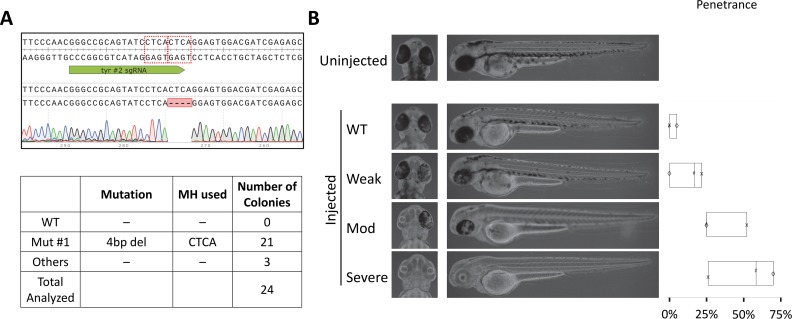
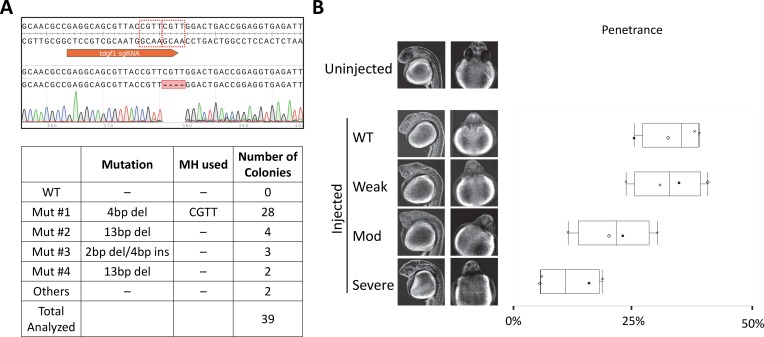
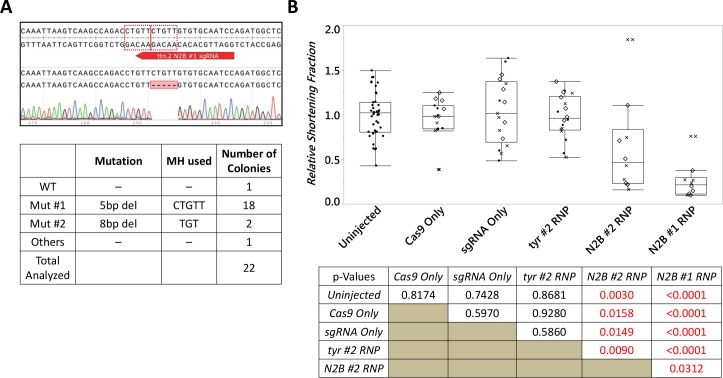
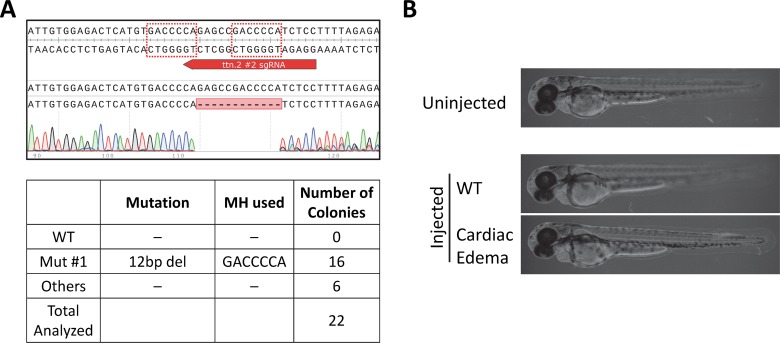
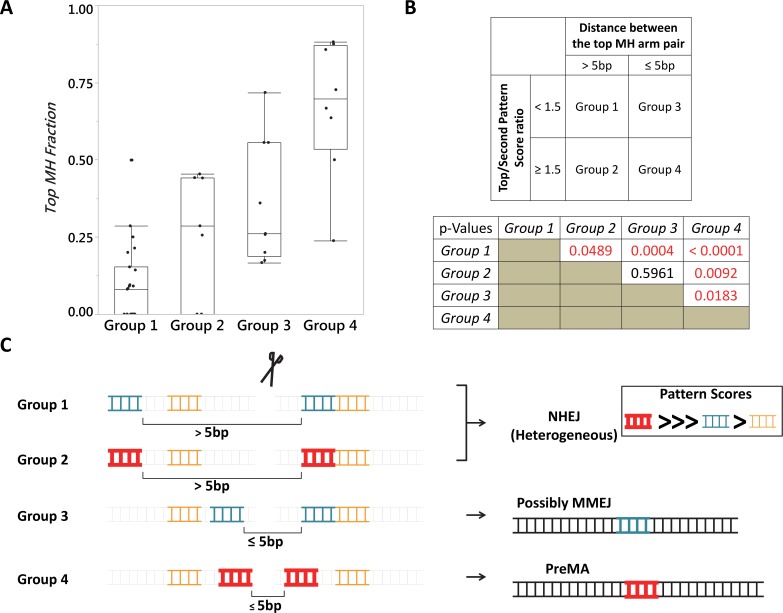
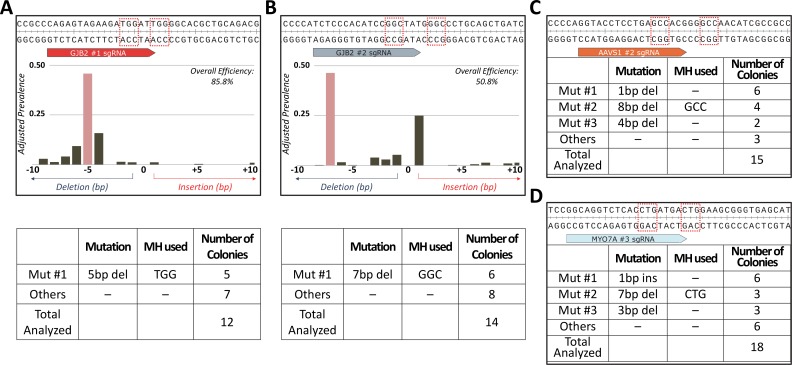
One key problem in precision genome editing is the unpredictable plurality of sequence outcomes at the site of targeted DNA double stranded breaks (DSBs). This is due to the typical activation of the versatile Non-homologous End Joining (NHEJ) pathway. Such unpredictability limits the utility of somatic gene editing for applications including gene therapy and functional genomics. For germline editing work, the accurate reproduction of the identical alleles using NHEJ is a labor intensive process. In this study, we propose Microhomology-mediated End Joining (MMEJ) as a viable solution for improving somatic sequence homogeneity in vivo, capable of generating a single predictable allele at high rates (56% ~ 86% of the entire mutant allele pool). Using a combined dataset from zebrafish (Danio rerio) in vivo and human HeLa cell in vitro, we identified specific contextual sequence determinants surrounding genomic DSBs for robust MMEJ pathway activation. We then applied our observation to prospectively design MMEJ-inducing sgRNAs against a variety of proof-of-principle genes and demonstrated high levels of mutant allele homogeneity. MMEJ-based DNA repair at these target loci successfully generated F0 mutant zebrafish embryos and larvae that faithfully recapitulated previously reported, recessive, loss-of-function phenotypes. We also tested the generalizability of our approach in cultured human cells. Finally, we provide a novel algorithm, MENTHU (http://genesculpt.org/menthu/), for improved and facile prediction of candidate MMEJ loci. We believe that this MMEJ-centric approach will have a broader impact on genome engineering and its applications. For example, whereas somatic mosaicism hinders efficient recreation of knockout mutant allele at base pair resolution via the standard NHEJ-based approach, we demonstrate that F0 founders transmitted the identical MMEJ allele of interest at high rates. Most importantly, the ability to directly dictate the reading frame of an endogenous target will have important implications for gene therapy applications in human genetic diseases.
Conflict of interest statement
The authors declare no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
