

Accurate classification of BRCA1 variants with saturation genome editing
- PMID: 30209399
- PMCID: PMC6181777
- DOI: 10.1038/s41586-018-0461-z
Accurate classification of BRCA1 variants with saturation genome editing
Abstract
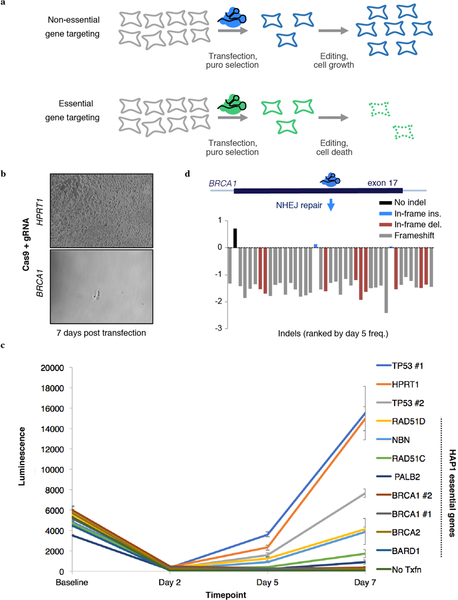
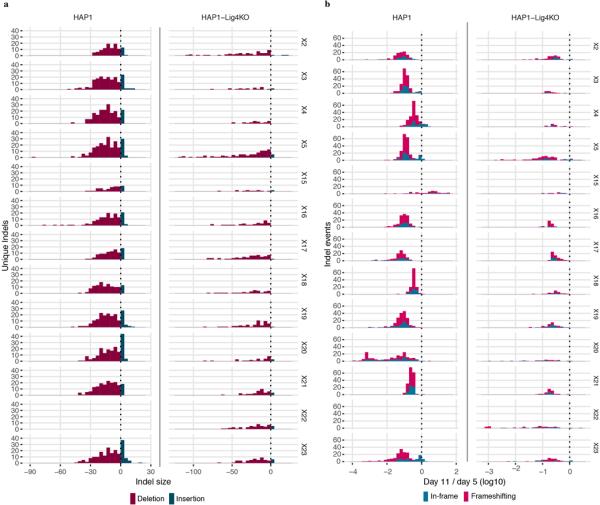
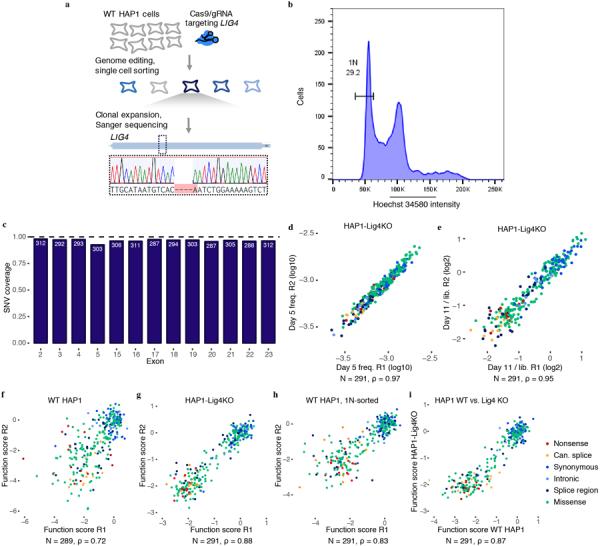
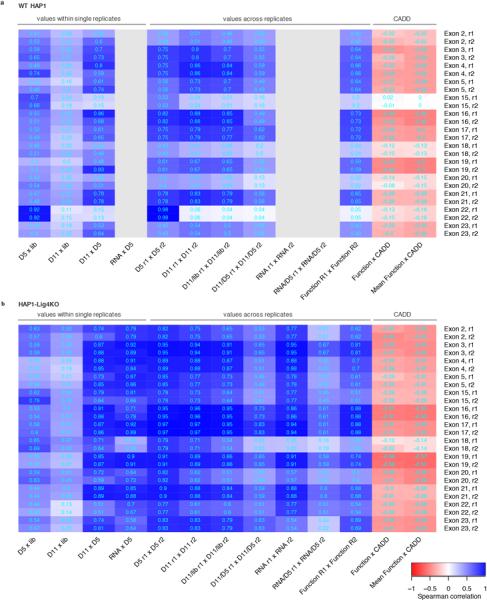
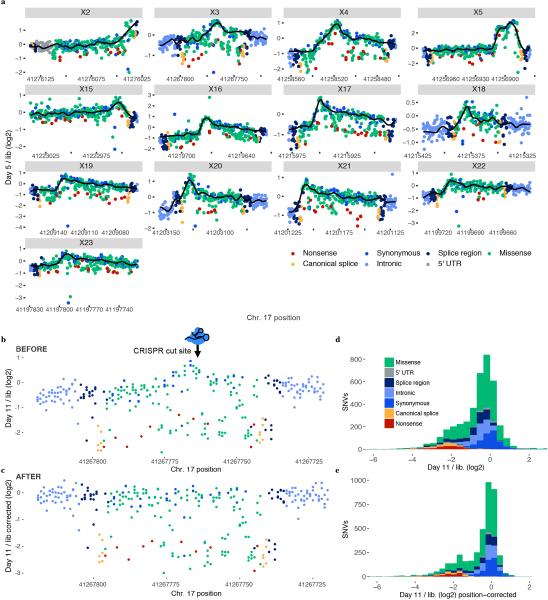
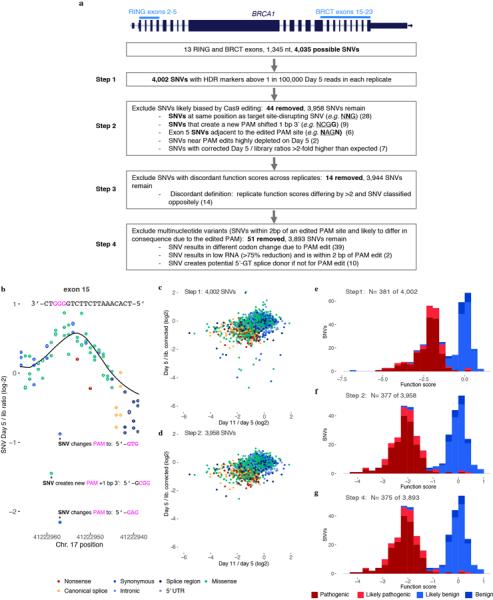
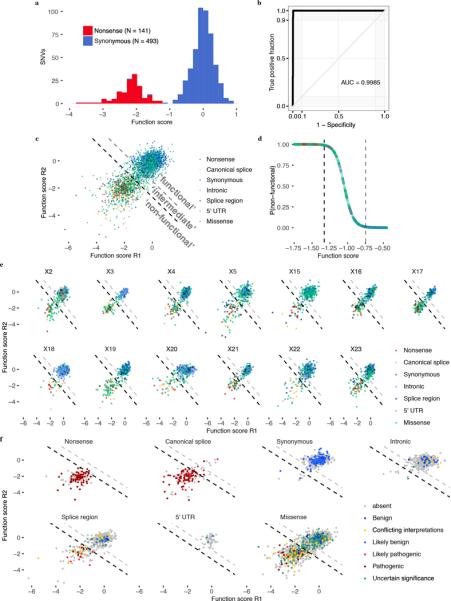
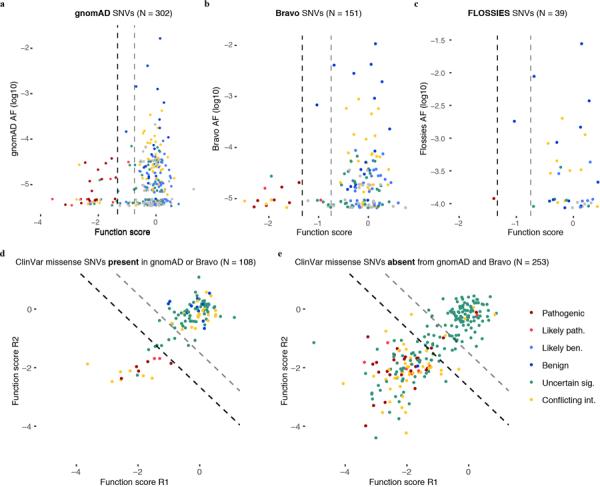
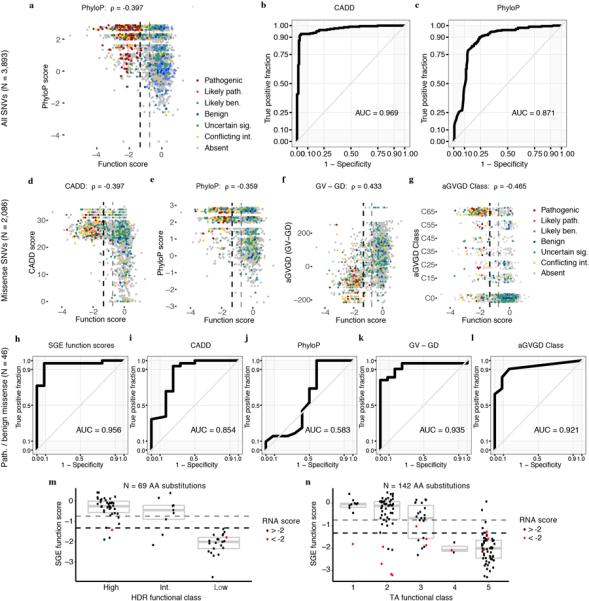
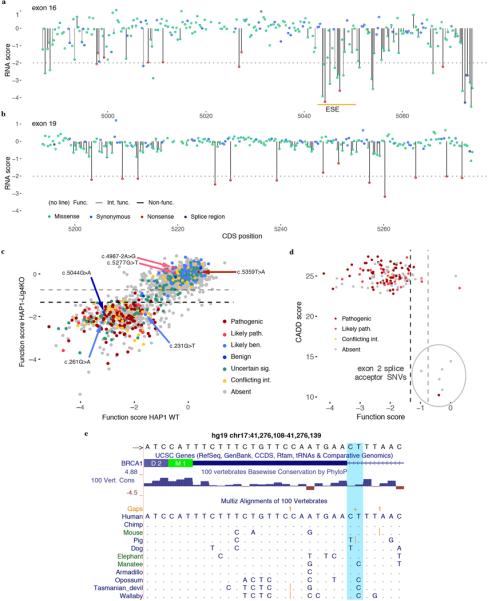
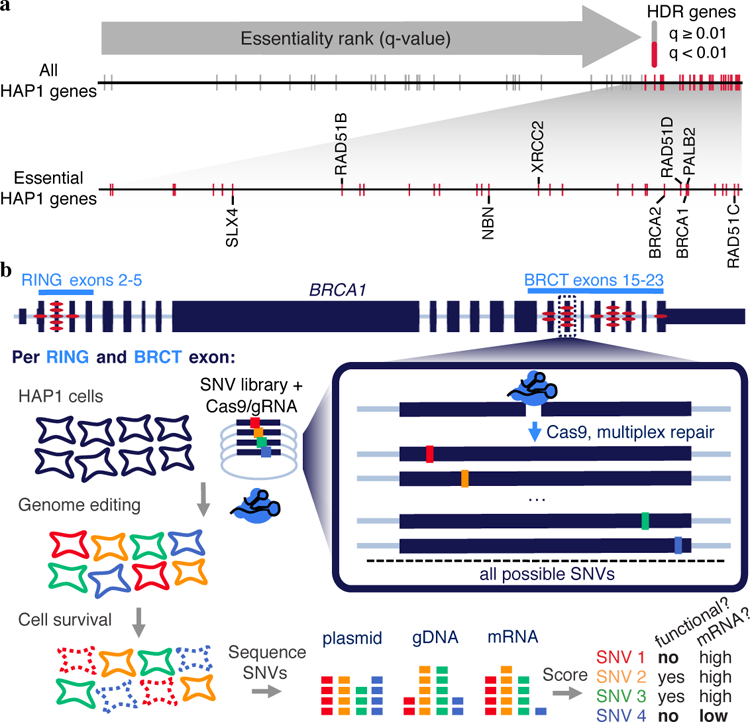
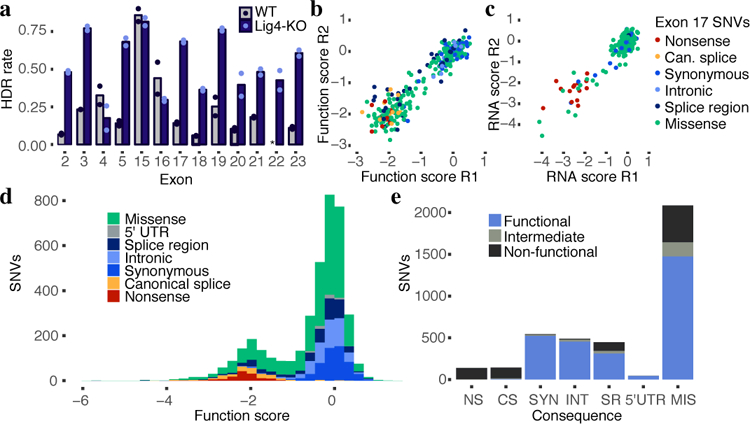
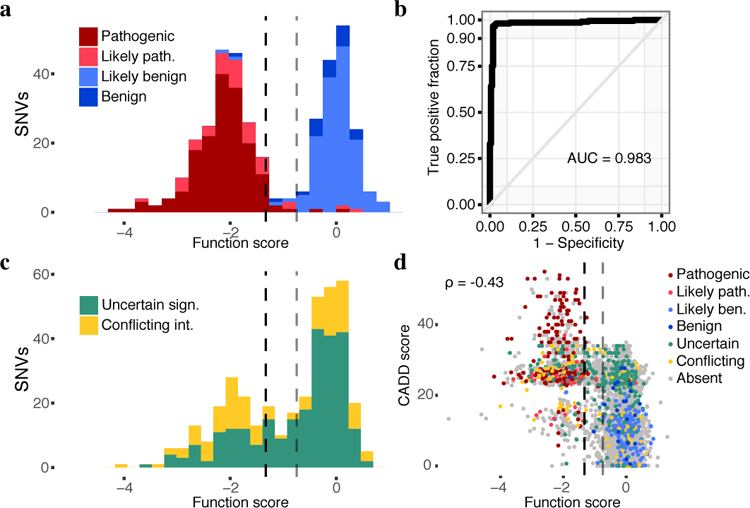
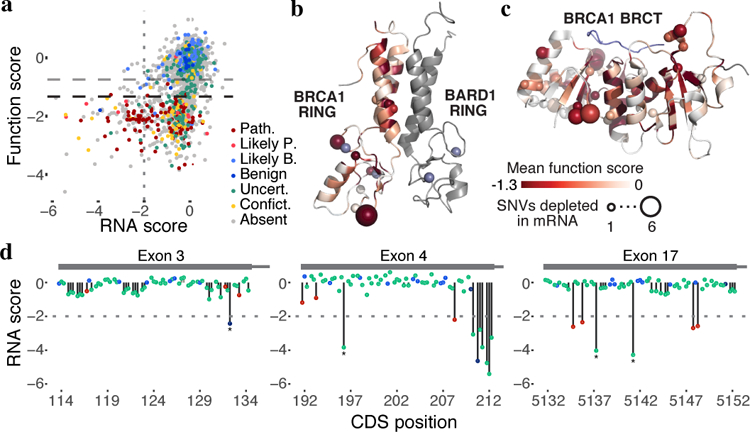
Variants of uncertain significance fundamentally limit the clinical utility of genetic information. The challenge they pose is epitomized by BRCA1, a tumour suppressor gene in which germline loss-of-function variants predispose women to breast and ovarian cancer. Although BRCA1 has been sequenced in millions of women, the risk associated with most newly observed variants cannot be definitively assigned. Here we use saturation genome editing to assay 96.5% of all possible single-nucleotide variants (SNVs) in 13 exons that encode functionally critical domains of BRCA1. Functional effects for nearly 4,000 SNVs are bimodally distributed and almost perfectly concordant with established assessments of pathogenicity. Over 400 non-functional missense SNVs are identified, as well as around 300 SNVs that disrupt expression. We predict that these results will be immediately useful for the clinical interpretation of BRCA1 variants, and that this approach can be extended to overcome the challenge of variants of uncertain significance in additional clinically actionable genes.
Figures

Comment in
-
Putting genetic variants to a fitness test.Nat Rev Genet. 2018 Nov;19(11):667. doi: 10.1038/s41576-018-0056-4. Nat Rev Genet. 2018. PMID: 30279525 No abstract available.
-
Gene editing reveals the effect of thousands of variants in a key cancer gene.Nature. 2018 Oct;562(7726):201-202. doi: 10.1038/d41586-018-06022-y. Nature. 2018. PMID: 30291308 No abstract available.
-
Interpreting the unknown.Nat Med. 2018 Oct;24(10):1491. doi: 10.1038/s41591-018-0227-z. Nat Med. 2018. PMID: 30297893 No abstract available.
-
Technique Pinpoints Oncogenic BRCA1 Mutations.Cancer Discov. 2018 Dec;8(12):OF8. doi: 10.1158/2159-8290.CD-NB2018-128. Epub 2018 Oct 12. Cancer Discov. 2018. PMID: 30314988
-
A pitfall in targeted Sanger sequencing of BRCA splicing variants in at-risk individuals.Pathol Res Pract. 2021 Jun;222:153456. doi: 10.1016/j.prp.2021.153456. Epub 2021 Apr 28. Pathol Res Pract. 2021. PMID: 33964677 No abstract available.
References
-
- Kuchenbaecker KB et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 317, 2402–2416 (2017). - PubMed
-
- Hall JM et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 250, 1684–1689 (1990). - PubMed
-
- Olopade OI & Artioli G Efficacy of risk-reducing salpingo-oophorectomy in women with BRCA-1 and BRCA-2 mutations. Breast J 10 Suppl 1, S5–9 (2004). - PubMed
-
- Rebbeck TR et al. Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J. Clin. Oncol 22, 1055–1062 (2004). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
