

Gene Therapy in Mouse Models of Deafness and Balance Dysfunction
- PMID: 30210291
- PMCID: PMC6123355
- DOI: 10.3389/fnmol.2018.00300
Gene Therapy in Mouse Models of Deafness and Balance Dysfunction
Abstract
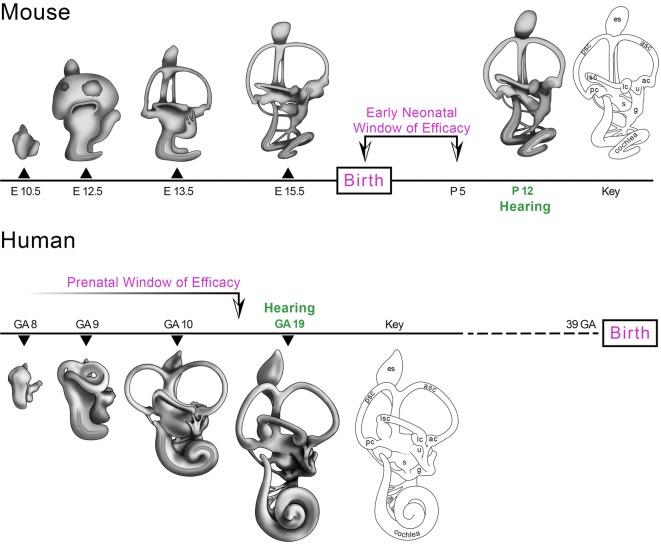
Therapeutic strategies to restore hearing and balance in mouse models of inner ear disease aim to rescue sensory function by gene replacement, augmentation, knock down or knock out. Modalities to achieve therapeutic effects have utilized virus-mediated transfer of wild type genes and small interfering ribonucleic acids; systemic and focal administration of antisense oligonucleotides (ASO) and designer small molecules; and lipid-mediated transfer of Cas 9 ribonucleoprotein (RNP) complexes. This work has established that gene or drug administration to the structurally and functionally immature, early neonatal mouse inner ear prior to hearing onset is a prerequisite for the most robust therapeutic responses. These observations may have significant implications for translating mouse inner ear gene therapies to patients. The human fetus hears by gestational week 19, suggesting that a corollary window of therapeutic efficacy closes early in the second trimester of pregnancy. We hypothesize that fetal therapeutics deployed prior to hearing onset may be the most effective approach to preemptively manage genetic mutations that cause deafness and vestibular dysfunction. We assert that gene therapy studies in higher vertebrate model systems with fetal hearing onset and a comparable acoustic range and sensitivity to that of humans are an essential step to safely and effectively translate murine gene therapies to the clinic.
Keywords: congenital deafness; fetal gene transfer; gene therapy; transuterine microinjection; window of therapeutic efficacy.
Figures
Similar articles
-
Fetal gene therapy and pharmacotherapy to treat congenital hearing loss and vestibular dysfunction.Hear Res. 2020 Sep 1;394:107931. doi: 10.1016/j.heares.2020.107931. Epub 2020 Mar 5. Hear Res. 2020. PMID: 32173115 Free PMC article. Review.
-
Fetal antisense oligonucleotide therapy for congenital deafness and vestibular dysfunction.Nucleic Acids Res. 2020 May 21;48(9):5065-5080. doi: 10.1093/nar/gkaa194. Nucleic Acids Res. 2020. PMID: 32249312 Free PMC article.
-
A Rapid, Cost-Effective Method to Prepare Recombinant Adeno-Associated Virus for Efficient Gene Transfer to the Developing Mouse Inner Ear.Methods Mol Biol. 2016;1427:43-57. doi: 10.1007/978-1-4939-3615-1_3. Methods Mol Biol. 2016. PMID: 27259920
-
Antisense oligonucleotides delivered to the amniotic cavity in utero modulate gene expression in the postnatal mouse.Nucleic Acids Res. 2016 Nov 16;44(20):9519-9529. doi: 10.1093/nar/gkw867. Epub 2016 Sep 28. Nucleic Acids Res. 2016. PMID: 27683224 Free PMC article.
-
Gene editing based hearing impairment research and therapeutics.Neurosci Lett. 2019 Sep 14;709:134326. doi: 10.1016/j.neulet.2019.134326. Epub 2019 Jun 10. Neurosci Lett. 2019. PMID: 31195050 Review.
Cited by
-
Strategies for non-viral vectors targeting organs beyond the liver.Nat Nanotechnol. 2024 Apr;19(4):428-447. doi: 10.1038/s41565-023-01563-4. Epub 2023 Dec 27. Nat Nanotechnol. 2024. PMID: 38151642 Review.
-
Engraftment of Human Stem Cell-Derived Otic Progenitors in the Damaged Cochlea.Mol Ther. 2019 Jun 5;27(6):1101-1113. doi: 10.1016/j.ymthe.2019.03.018. Epub 2019 Apr 2. Mol Ther. 2019. PMID: 31005598 Free PMC article.
-
Inner ear organoids: new tools to understand neurosensory cell development, degeneration and regeneration.Development. 2019 Sep 2;146(17):dev177188. doi: 10.1242/dev.177188. Development. 2019. PMID: 31477580 Free PMC article. Review.
-
Fetal gene therapy and pharmacotherapy to treat congenital hearing loss and vestibular dysfunction.Hear Res. 2020 Sep 1;394:107931. doi: 10.1016/j.heares.2020.107931. Epub 2020 Mar 5. Hear Res. 2020. PMID: 32173115 Free PMC article. Review.
-
Molecular Characterization of Subdomain Specification of Cochlear Duct Based on Foxg1 and Gata3.Int J Mol Sci. 2024 Nov 26;25(23):12700. doi: 10.3390/ijms252312700. Int J Mol Sci. 2024. PMID: 39684410 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources