

Toll-like Receptor 3 Is a Therapeutic Target for Pulmonary Hypertension
- PMID: 30211629
- PMCID: PMC6353001
- DOI: 10.1164/rccm.201707-1370OC
Toll-like Receptor 3 Is a Therapeutic Target for Pulmonary Hypertension
Abstract
Rationale: Pulmonary arterial hypertension (PAH) is characterized by vascular cell proliferation and endothelial cell apoptosis. TLR3 (Toll-like receptor 3) is a receptor for double-stranded RNA and has been recently implicated in vascular protection.
Objectives: To study the expression and role of TLR3 in PAH and to determine whether a TLR3 agonist reduces pulmonary hypertension in preclinical models.
Methods: Lung tissue and endothelial cells from patients with PAH were investigated by polymerase chain reaction, immunofluorescence, and apoptosis assays. TLR3-/- and TLR3+/+ mice were exposed to chronic hypoxia and SU5416. Chronic hypoxia or chronic hypoxia/SU5416 rats were treated with the TLR3 agonist polyinosinic/polycytidylic acid (Poly[I:C]).
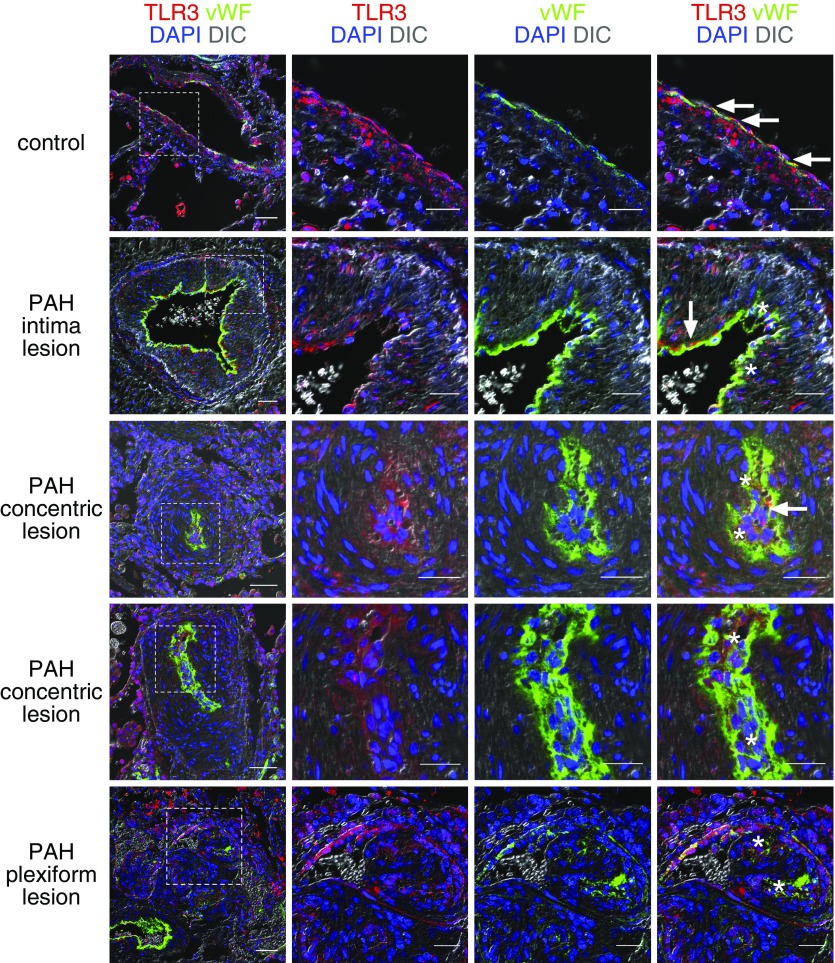
Measurements and main results: TLR3 expression was reduced in PAH patient lung tissue and endothelial cells, and TLR3-/- mice exhibited more severe pulmonary hypertension following exposure to chronic hypoxia/SU5416. TLR3 knockdown promoted double-stranded RNA signaling via other intracellular RNA receptors in endothelial cells. This was associated with greater susceptibility to apoptosis, a known driver of pulmonary vascular remodeling. Poly(I:C) increased TLR3 expression via IL-10 in rat endothelial cells. In vivo, high-dose Poly(I:C) reduced pulmonary hypertension in both rat models in proof-of-principle experiments. In addition, Poly(I:C) also reduced right ventricular failure in established pulmonary hypertension.
Conclusions: Our work identifies a novel role for TLR3 in PAH based on the findings that reduced expression of TLR3 contributes to endothelial apoptosis and pulmonary vascular remodeling.
Keywords: apoptosis; double-stranded RNA; endothelial cell; pulmonary hypertension; toll-like receptor 3.
Figures





Comment in
-
An RNA Sensor Protects against Pulmonary Hypertension.Am J Respir Crit Care Med. 2019 Jan 15;199(2):138-140. doi: 10.1164/rccm.201807-1363ED. Am J Respir Crit Care Med. 2019. PMID: 30252495 No abstract available.
References
-
- Macchia A, Marchioli R, Tognoni G, Scarano M, Marfisi R, Tavazzi L, et al. Systematic review of trials using vasodilators in pulmonary arterial hypertension: why a new approach is needed. Am Heart J. 2010;159:245–257. - PubMed
-
- Chaudhary KR, Taha M, Cadete VJ, Godoy RS, Stewart DJ. Proliferative versus degenerative paradigms in pulmonary arterial hypertension: have we put the cart before the horse? Circ Res. 2017;120:1237–1239. - PubMed
-
- Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. 2005;19:1178–1180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- PG/18/1/33430/BHF_/British Heart Foundation/United Kingdom
- P01 HL103455/HL/NHLBI NIH HHS/United States
- R03 HL114816/HL/NHLBI NIH HHS/United States
- R01 HL139881/HL/NHLBI NIH HHS/United States
- FS/18/13/33281/BHF_/British Heart Foundation/United Kingdom
- PG/11/46/28979/BHF_/British Heart Foundation/United Kingdom
- R24 HL123767/HL/NHLBI NIH HHS/United States
- P30 CA016059/CA/NCI NIH HHS/United States
- R21 HL123044/HL/NHLBI NIH HHS/United States
- FS/13/48/30453/BHF_/British Heart Foundation/United Kingdom
- PG/19/41/34426/BHF_/British Heart Foundation/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
