

Growth-restricting effects of siRNA transfections: a largely deterministic combination of off-target binding and hybridization-independent competition
- PMID: 30215772
- PMCID: PMC6182159
- DOI: 10.1093/nar/gky798
Growth-restricting effects of siRNA transfections: a largely deterministic combination of off-target binding and hybridization-independent competition
Abstract
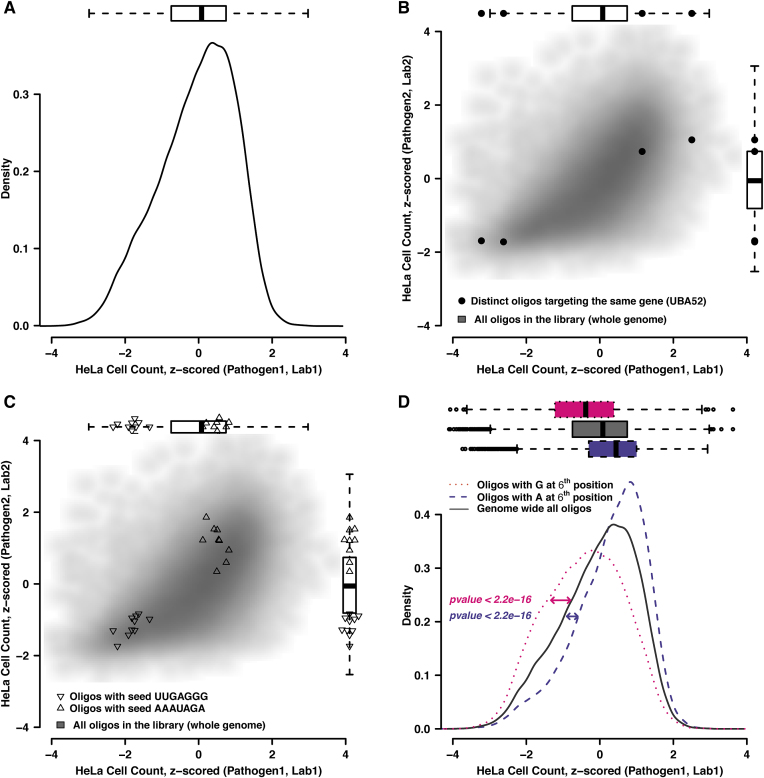
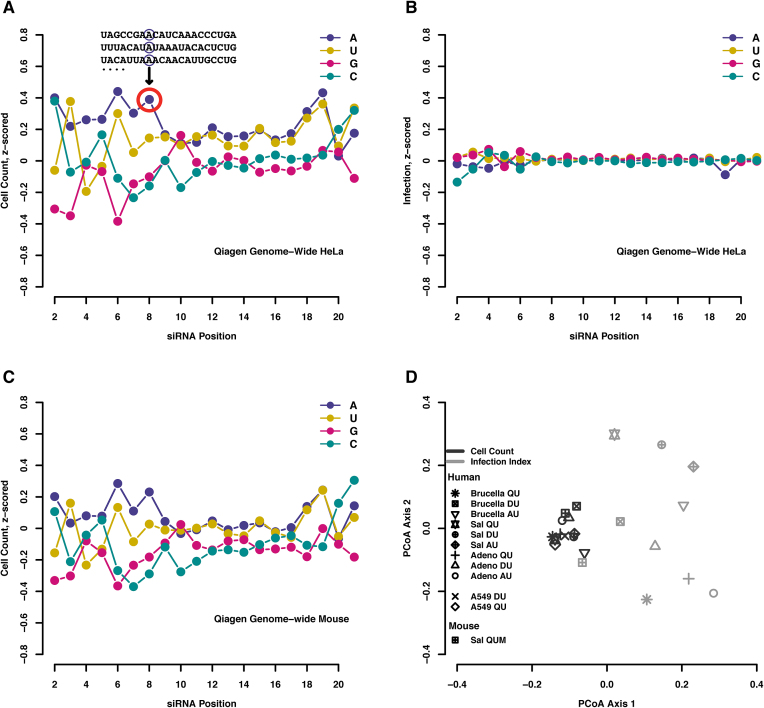
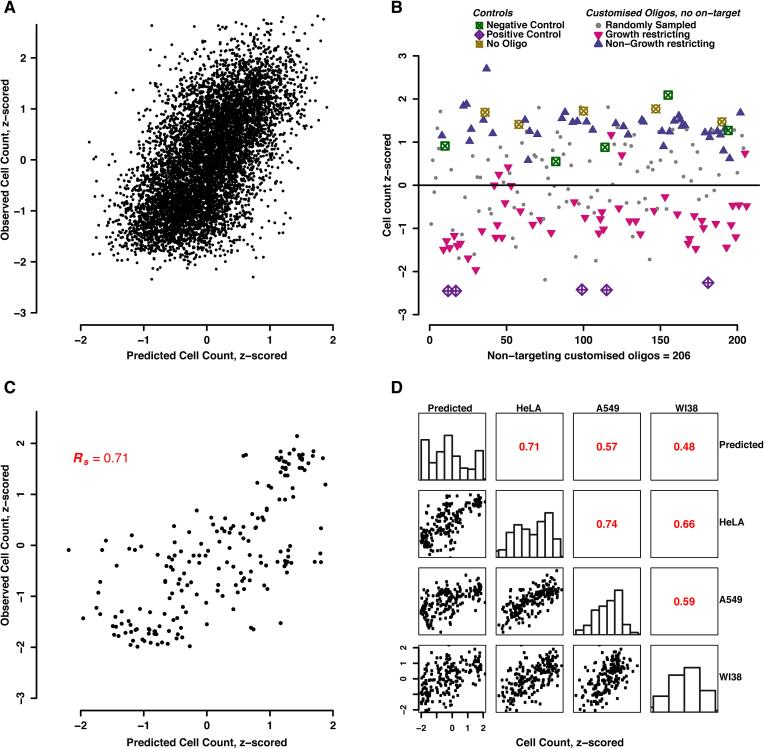
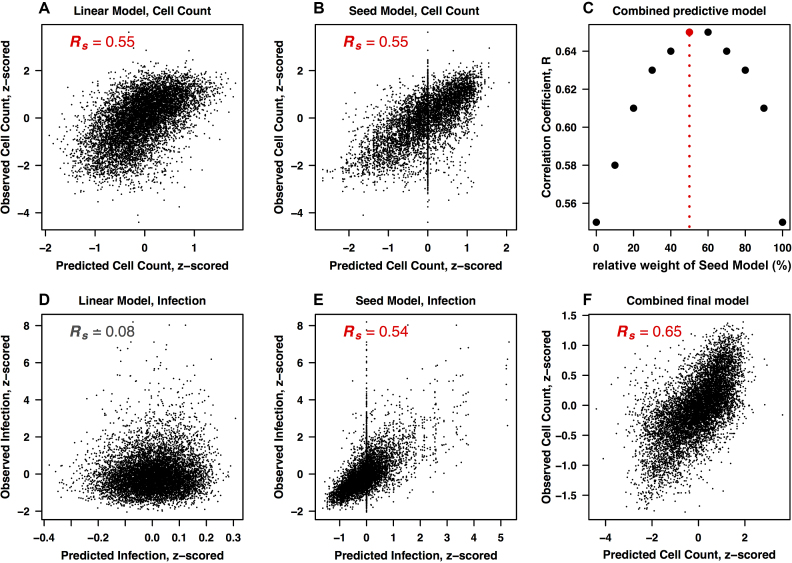
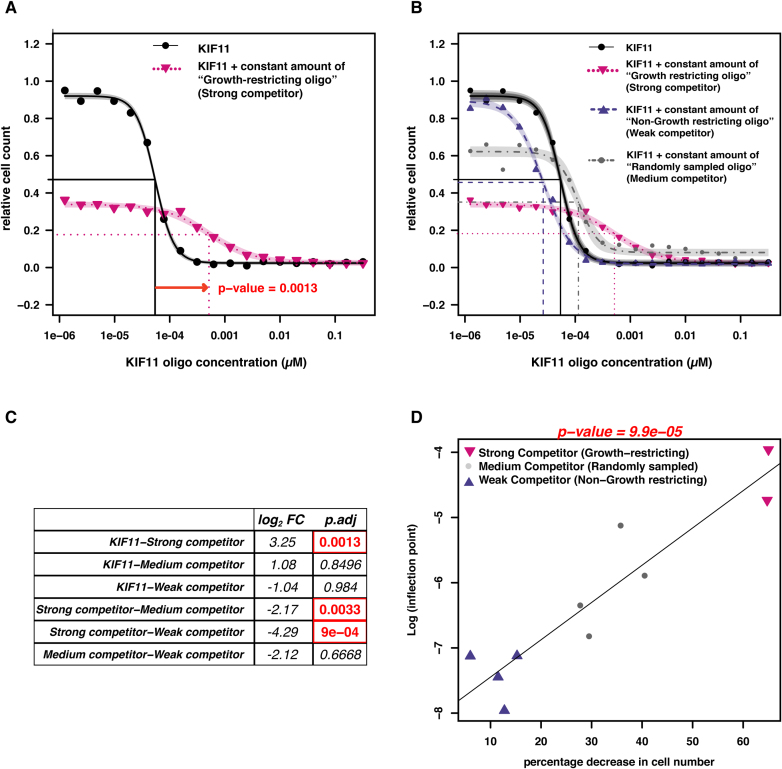
Perturbation of gene expression by means of synthetic small interfering RNAs (siRNAs) is a powerful way to uncover gene function. However, siRNA technology suffers from sequence-specific off-target effects and from limitations in knock-down efficiency. In this study, we assess a further problem: unintended effects of siRNA transfections on cellular fitness/proliferation. We show that the nucleotide compositions of siRNAs at specific positions have reproducible growth-restricting effects on mammalian cells in culture. This is likely distinct from hybridization-dependent off-target effects, since each nucleotide residue is seen to be acting independently and additively. The effect is robust and reproducible across different siRNA libraries and also across various cell lines, including human and mouse cells. Analyzing the growth inhibition patterns in correlation to the nucleotide sequence of the siRNAs allowed us to build a predictor that can estimate growth-restricting effects for any arbitrary siRNA sequence. Competition experiments with co-transfected siRNAs further suggest that the growth-restricting effects might be linked to an oversaturation of the cellular miRNA machinery, thus disrupting endogenous miRNA functions at large. We caution that competition between siRNA molecules could complicate the interpretation of double-knockdown or epistasis experiments, and potential interactions with endogenous miRNAs can be a factor when assaying cell growth or viability phenotypes.
Figures
References
-
- Taylor J., Woodcock S.. A perspective on the future of High-Throughput RNAi screening: will CRISPR cut out the competition or can RNAi help guide the way?. J. Biomol. Screen. 2015; 20:1040–1051. - PubMed
-
- Mello C.C., Conte D. Jr. Revealing the world of RNA interference. Nature. 2004; 431:338–342. - PubMed
-
- Meister G., Tuschl T.. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004; 431:343–349. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
