

Lysosomes Mediate Benefits of Intermittent Fasting in Cardiometabolic Disease: The Janitor Is the Undercover Boss
- PMID: 30215867
- PMCID: PMC6423516
- DOI: 10.1002/cphy.c180005
Lysosomes Mediate Benefits of Intermittent Fasting in Cardiometabolic Disease: The Janitor Is the Undercover Boss
Abstract
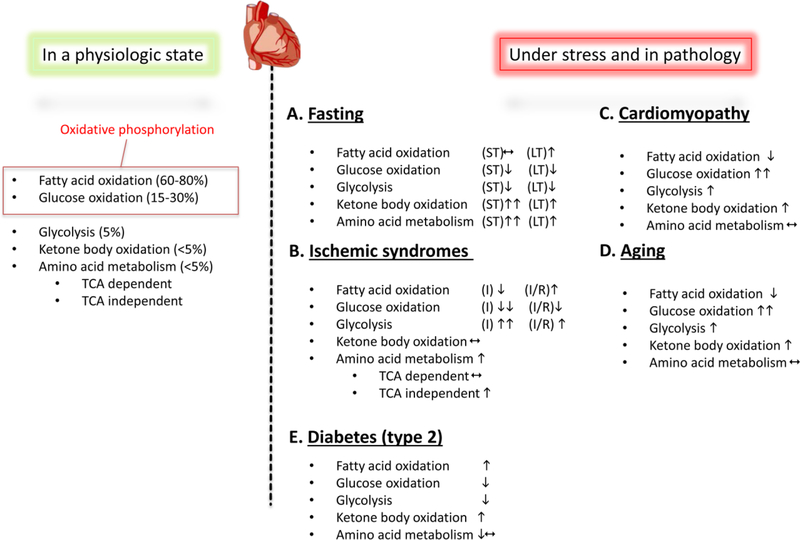
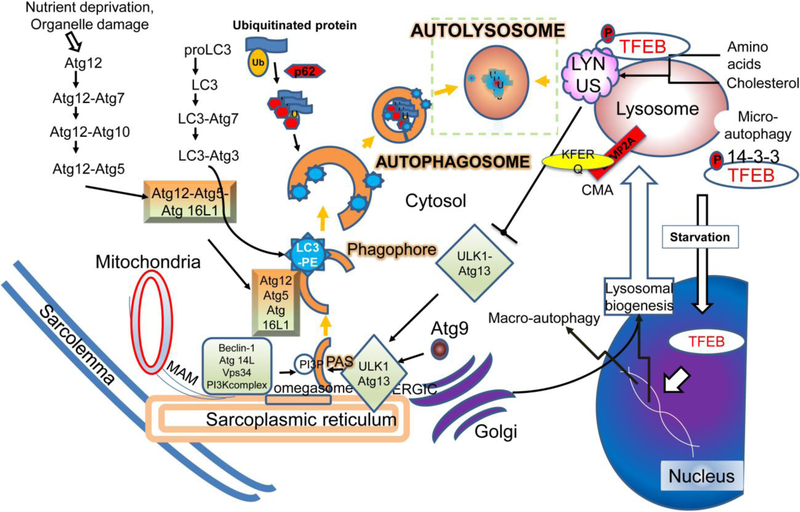
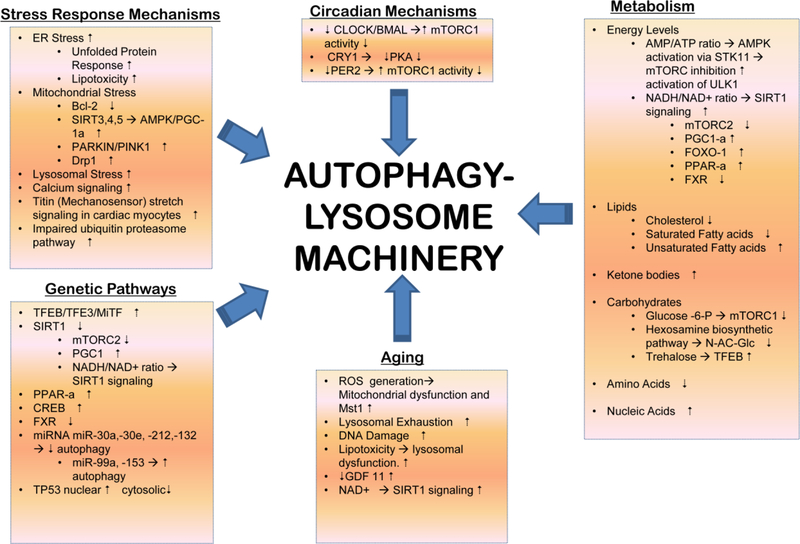
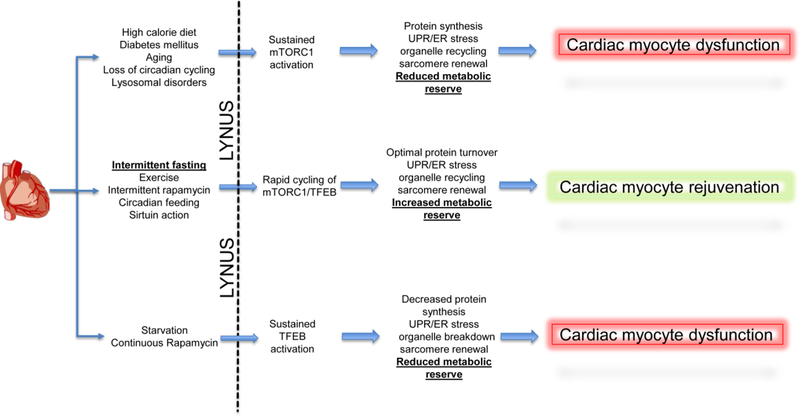
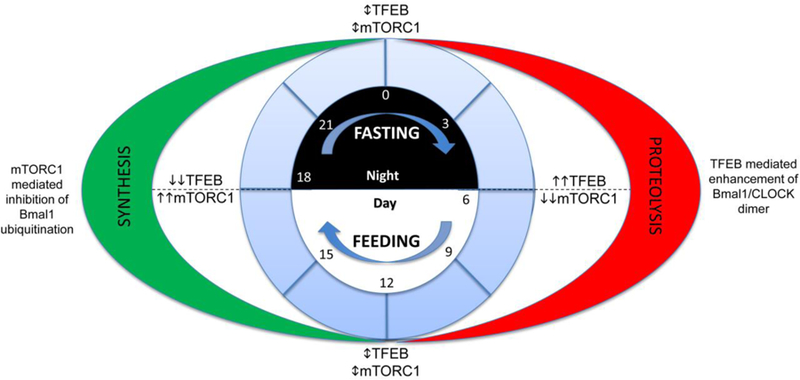
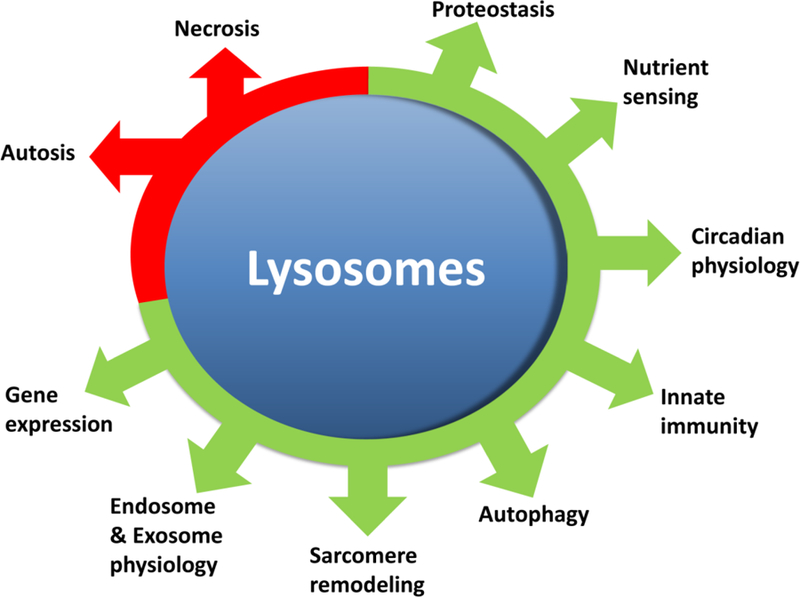
Adaptive responses that counter starvation have evolved over millennia to permit organismal survival, including changes at the level of individual organelles, cells, tissues, and organ systems. In the past century, a shift has occurred away from disease caused by insufficient nutrient supply toward overnutrition, leading to obesity and diabetes, atherosclerosis, and cardiometabolic disease. The burden of these diseases has spurred interest in fasting strategies that harness physiological responses to starvation, thus limiting tissue injury during metabolic stress. Insights gained from animal and human studies suggest that intermittent fasting and chronic caloric restriction extend lifespan, decrease risk factors for cardiometabolic and inflammatory disease, limit tissue injury during myocardial stress, and activate a cardioprotective metabolic program. Acute fasting activates autophagy, an intricately orchestrated lysosomal degradative process that sequesters cellular constituents for degradation, and is critical for cardiac homeostasis during fasting. Lysosomes are dynamic cellular organelles that function as incinerators to permit autophagy, as well as degradation of extracellular material internalized by endocytosis, macropinocytosis, and phagocytosis. The last decade has witnessed an explosion of knowledge that has shaped our understanding of lysosomes as central regulators of cellular metabolism and the fasting response. Intriguingly, lysosomes also store nutrients for release during starvation; and function as a nutrient sensing organelle to couple activation of mammalian target of rapamycin to nutrient availability. This article reviews the evidence for how the lysosome, in the guise of a janitor, may be the "undercover boss" directing cellular processes for beneficial effects of intermittent fasting and restoring homeostasis during feast and famine. © 2018 American Physiological Society. Compr Physiol 8:1639-1667, 2018.
Copyright © 2018 American Physiological Society. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
