

Ambiguous Effects of Autophagy Activation Following Hypoperfusion/Ischemia
- PMID: 30217100
- PMCID: PMC6163197
- DOI: 10.3390/ijms19092756
Ambiguous Effects of Autophagy Activation Following Hypoperfusion/Ischemia
Abstract
Autophagy primarily works to counteract nutrient deprivation that is strongly engaged during starvation and hypoxia, which happens in hypoperfusion. Nonetheless, autophagy is slightly active even in baseline conditions, when it is useful to remove aged proteins and organelles. This is critical when the mitochondria and/or proteins are damaged by toxic stimuli. In the present review, we discuss to that extent the recruitment of autophagy is beneficial in counteracting brain hypoperfusion or, vice-versa, its overactivity may per se be detrimental for cell survival. While analyzing these opposite effects, it turns out that the autophagy activity is likely not to be simply good or bad for cell survival, but its role varies depending on the timing and amount of autophagy activation. This calls for the need for an appropriate autophagy tuning to guarantee a beneficial effect on cell survival. Therefore, the present article draws a theoretical pattern of autophagy activation, which is hypothesized to define the appropriate timing and intensity, which should mirrors the duration and severity of brain hypoperfusion. The need for a fine tuning of the autophagy activation may explain why confounding outcomes occur when autophagy is studied using a rather simplistic approach.
Keywords: autophagy; brain ischemia; cerebral blood flow; hypoxia; mitophagy; neurodegeneration; starvation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
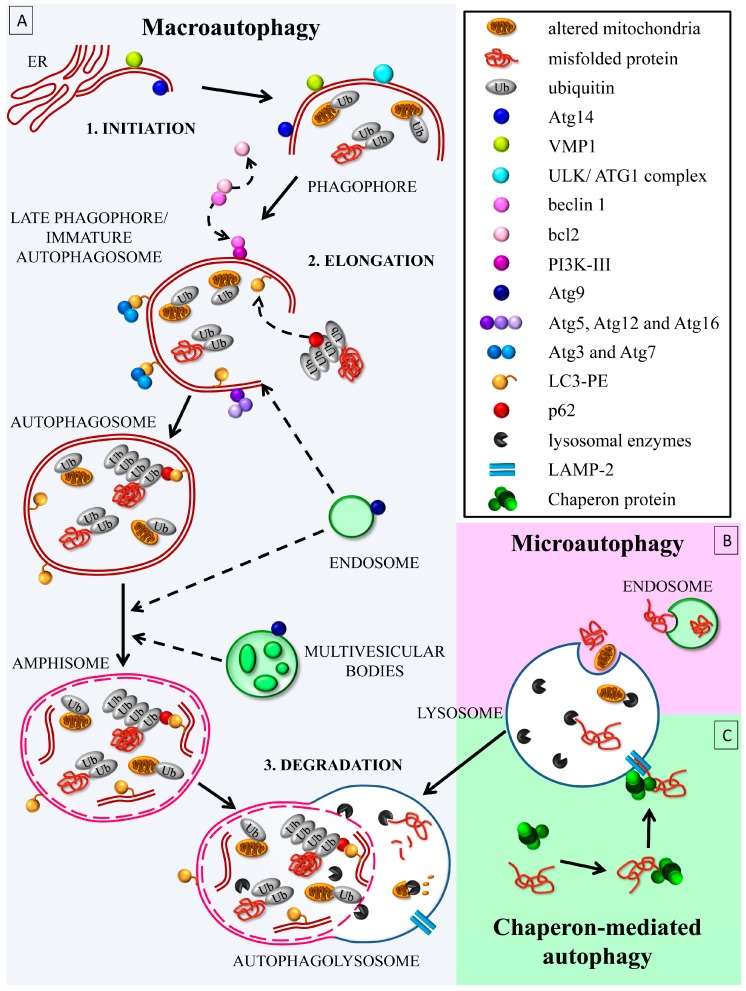
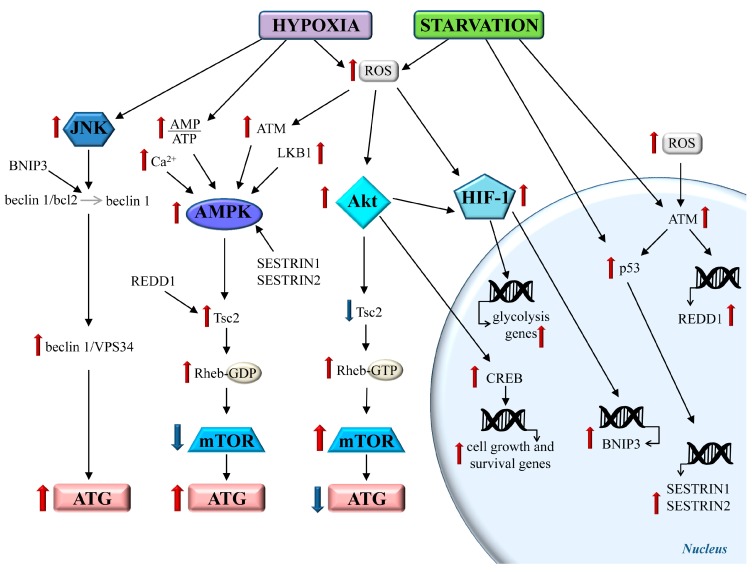
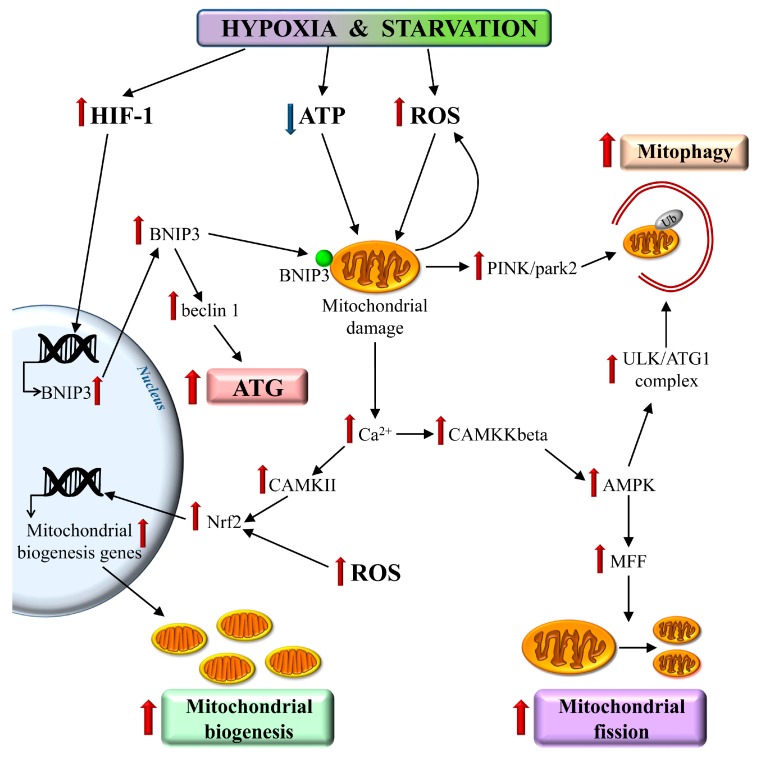
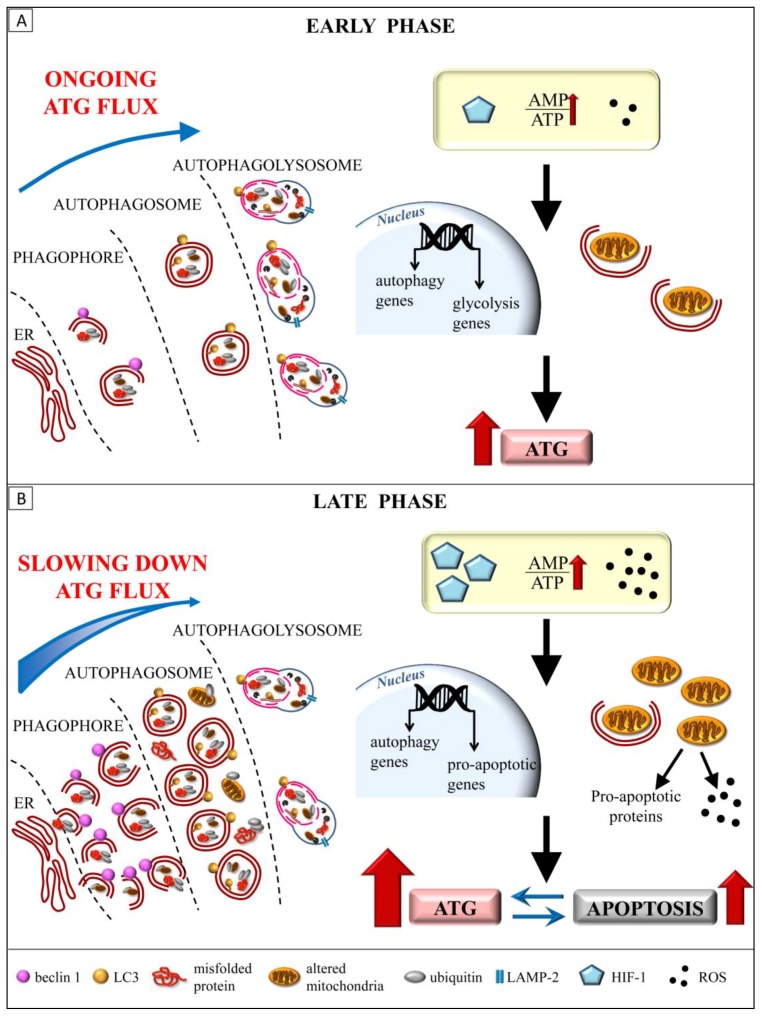
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
