

Cryptic sequence features in the active postmortem transcriptome
- PMID: 30217147
- PMCID: PMC6137749
- DOI: 10.1186/s12864-018-5042-x
Cryptic sequence features in the active postmortem transcriptome
Abstract
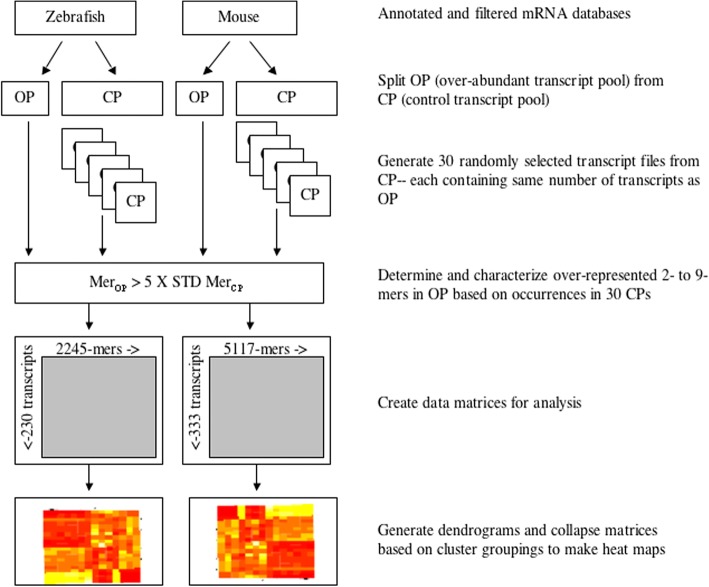
Background: Our previous study found that more than 500 transcripts significantly increased in abundance in the zebrafish and mouse several hours to days postmortem relative to live controls. The current literature suggests that most mRNAs are post-transcriptionally regulated in stressful conditions. We rationalized that the postmortem transcripts must contain sequence features (3- to 9- mers) that are unique from those in the rest of the transcriptome and that these features putatively serve as binding sites for proteins and/or non-coding RNAs involved in post-transcriptional regulation.
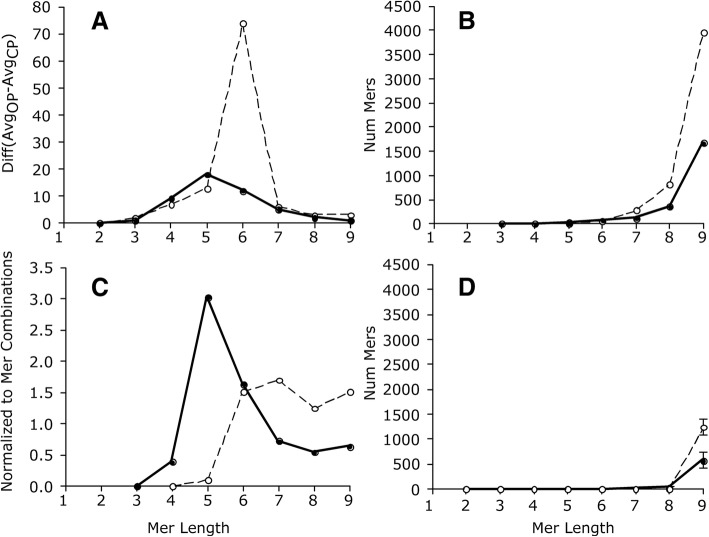
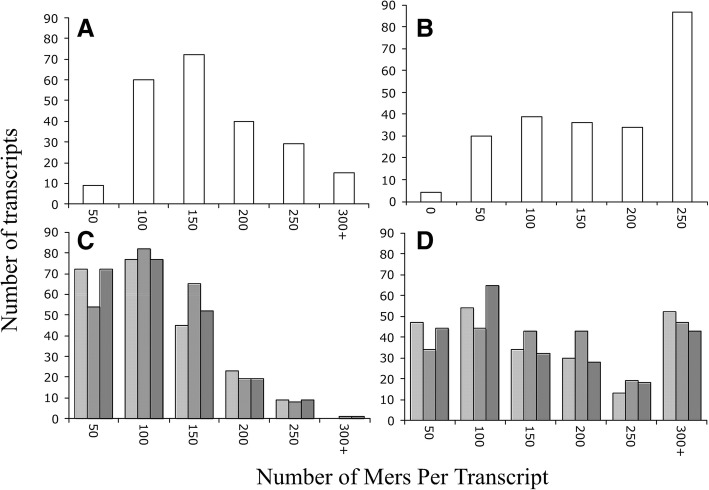
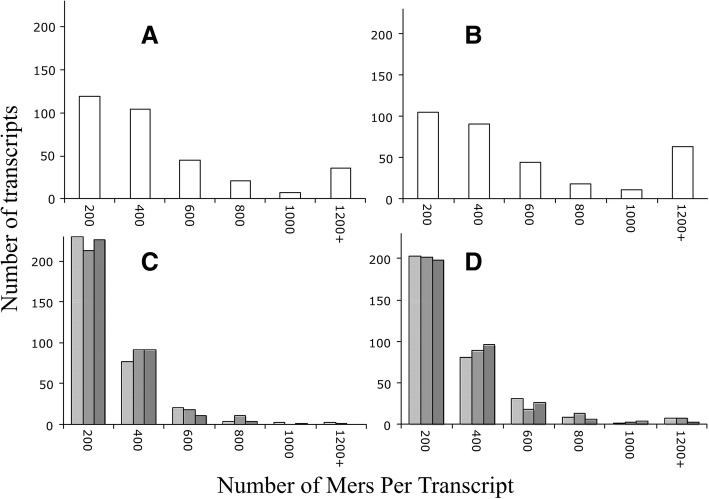
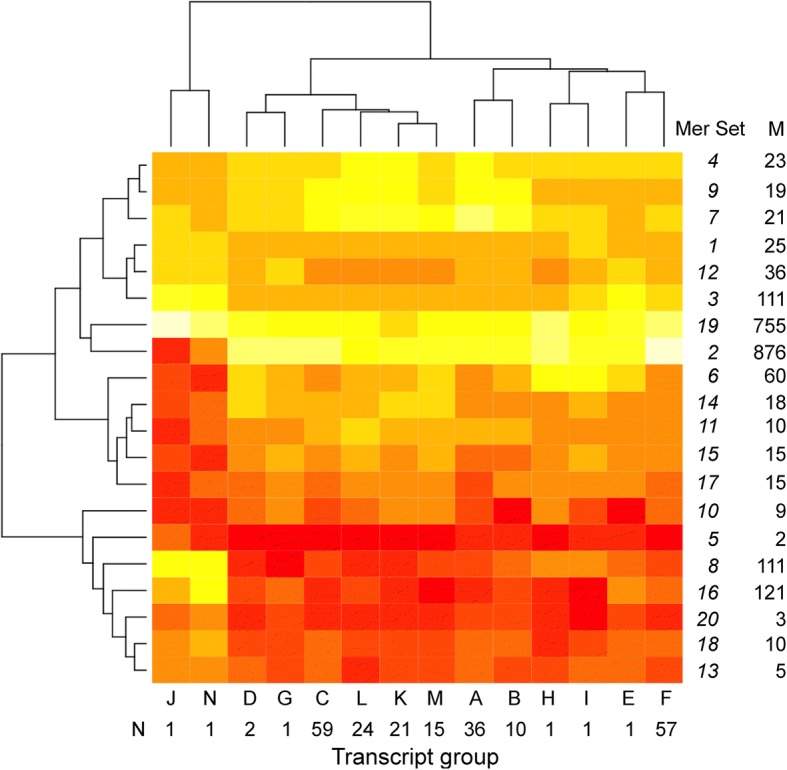
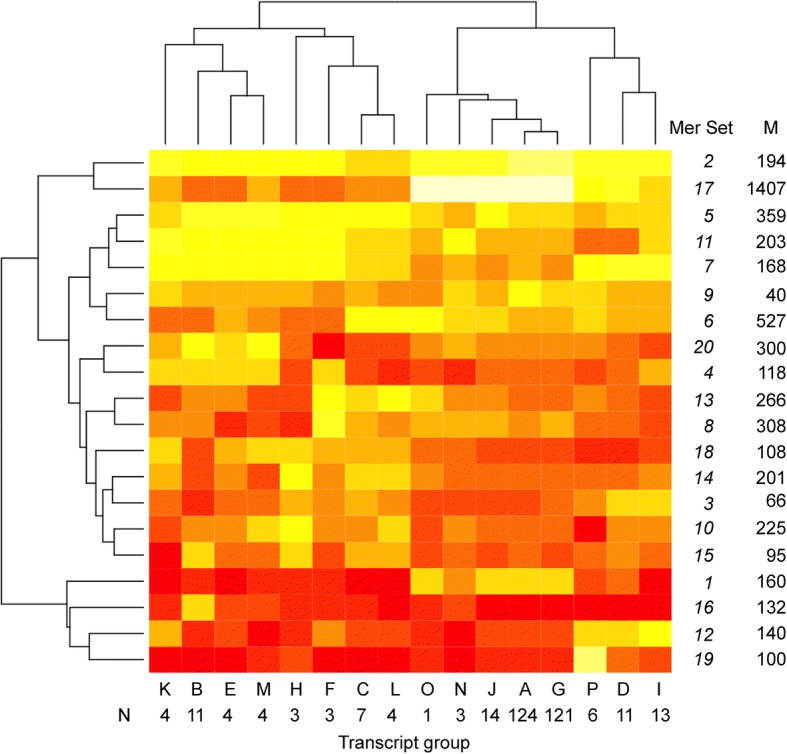
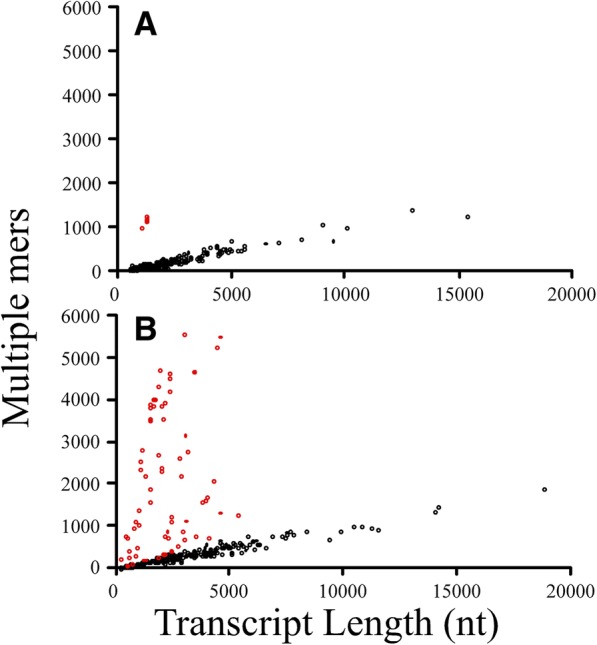
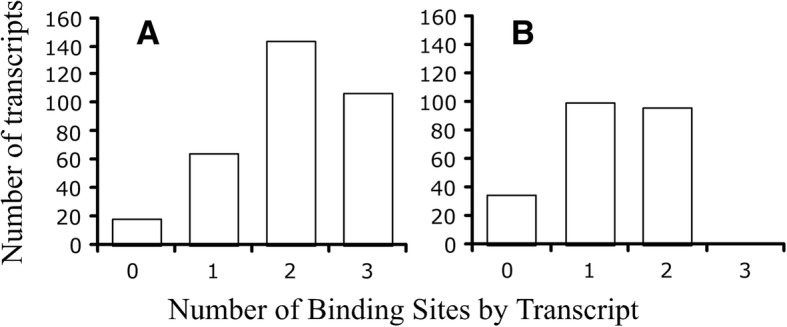
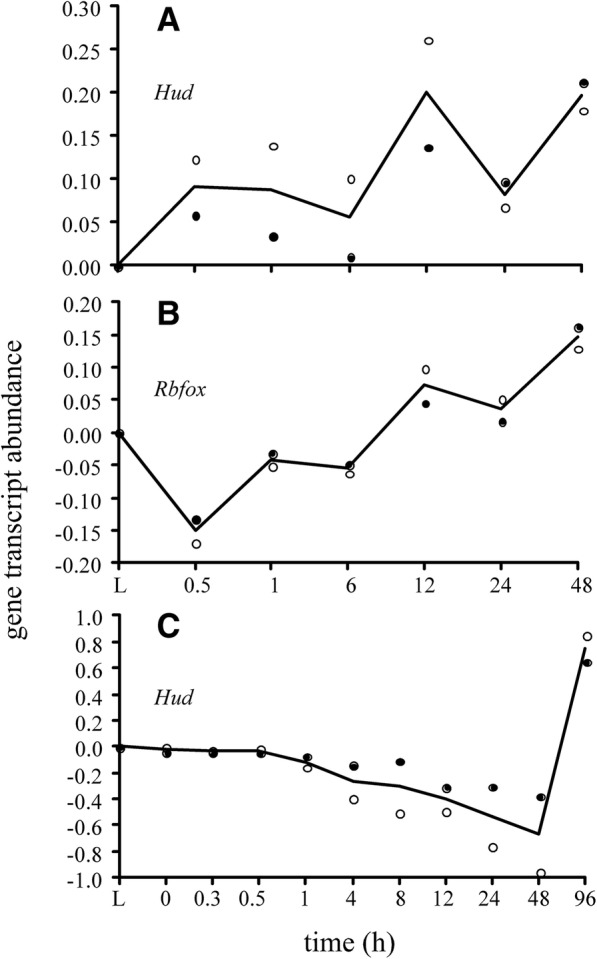
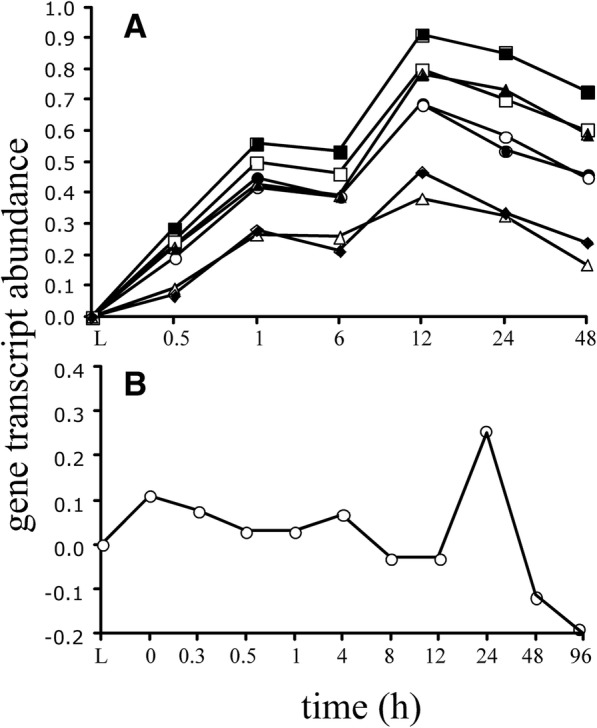
Results: We identified 5117 and 2245 over-represented sequence features in the mouse and zebrafish, respectively, which represents less than 1.5% of all possible features. Some of these features were disproportionately distributed along the transcripts with high densities in the 3' untranslated regions of the zebrafish (0.3 mers/nt) and the open reading frames of the mouse (0.6 mers/nt). Yet, the highest density (2.3 mers/nt) occurred in the open reading frames of 11 mouse transcripts that lacked 3' or 5' untranslated regions. These results suggest the transcripts with high density of features might serve as 'molecular sponges' that sequester RNA binding proteins and/or microRNAs, and thus indirectly increase the stability and gene expression of other transcripts. In addition, some of the features were identified as binding sites for Rbfox and Hud proteins that are also involved in increasing transcript stability and gene expression.
Conclusions: Our results are consistent with the hypothesis that transcripts involved in responding to extreme stress, such as organismal death, have sequence features that make them different from the rest of the transcriptome. Some of these features serve as putative binding sites for proteins and non-coding RNAs that determine transcript stability and fate. A small number of the transcripts have high density sequence features, which are presumably involved in sequestering RNA binding proteins and microRNAs and thus preventing regulatory interactions among other transcripts. Our results provide baseline data on post-transcriptional regulation in stressful conditions that has implications for regulation in disease, starvation, and cancer.
Keywords: 3’UTR; 5’UTR; Chaos game representation; Molecular sponge; Motifs; Mouse; ORFs; Post-transcriptional regulation; Postmortem gene expression; Sequence features; Stress response; Zebrafish.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable, all data publicly available.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Ferreira PG, Muñoz-Aguirre M, Reverter F, Sá Godinho CP, Sousa A, Amadoz A, Sodaei R, Hidalgo MR, Pervouchine, Dmitri, Carbonell-Caballero J, Nurtdinov R, Breschi A, Amador R, Oliveira P, Çubuk C, Curado J, Aguet F, Oliveira C, Dopazo J, Sammeth M, Ardlie KG, Guigó R. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat Commun. 2018;490:2041–1723. doi: 10.1038/s41467-017-02772-x. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
