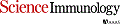Molecular diversification of regulatory T cells in nonlymphoid tissues
- PMID: 30217811
- PMCID: PMC6219455
- DOI: 10.1126/sciimmunol.aat5861
Molecular diversification of regulatory T cells in nonlymphoid tissues
Abstract
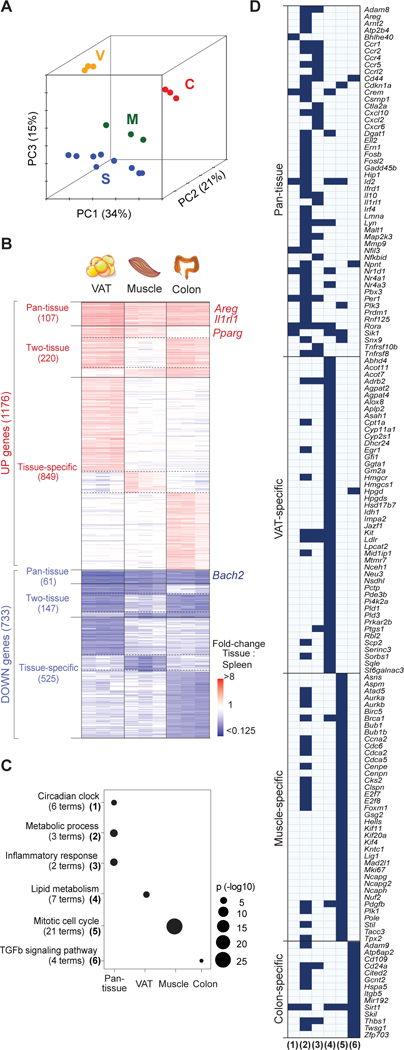
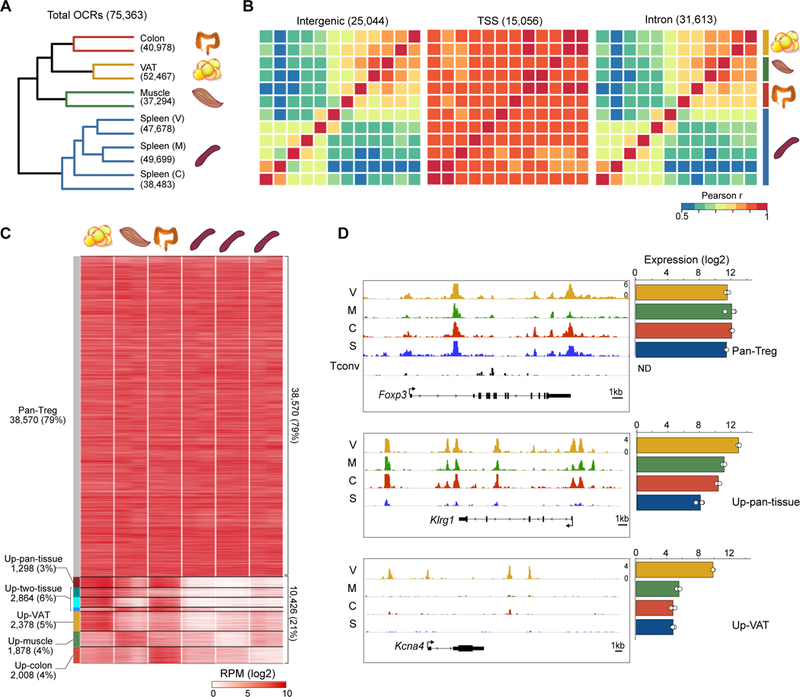
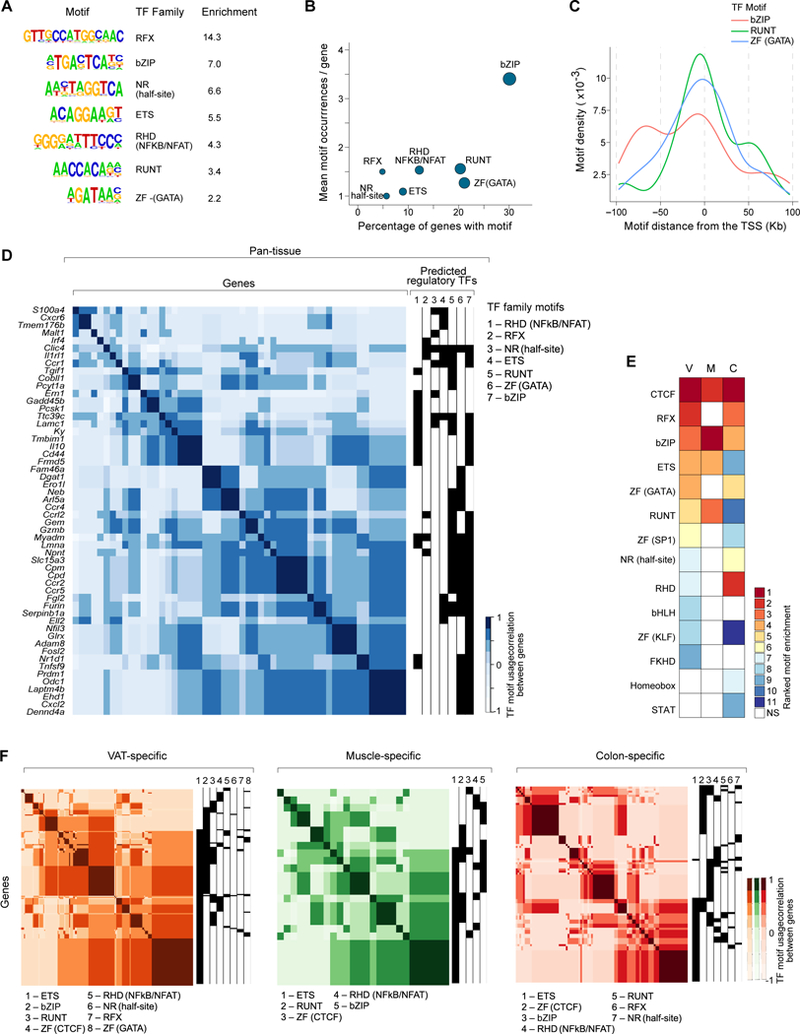
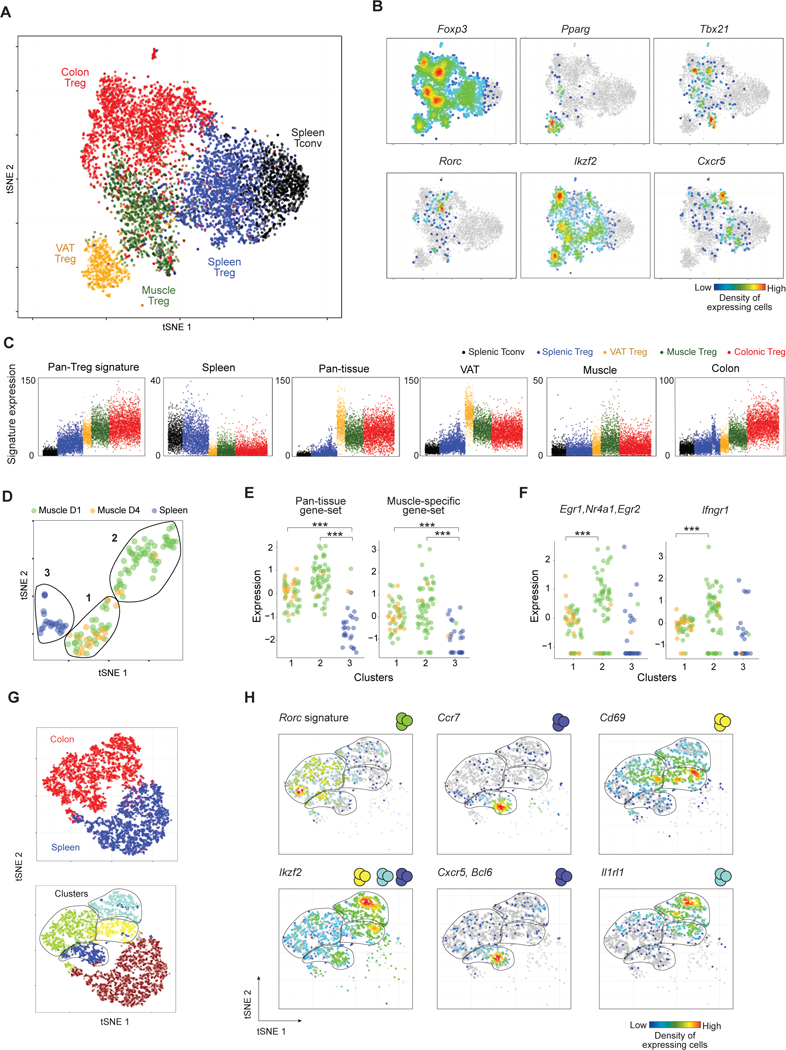
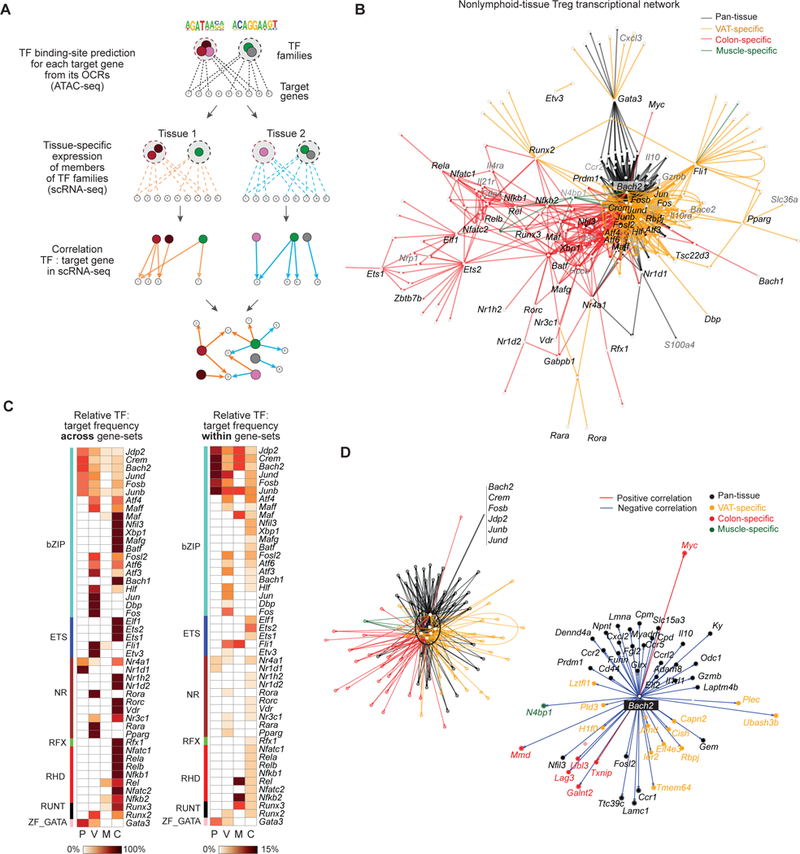
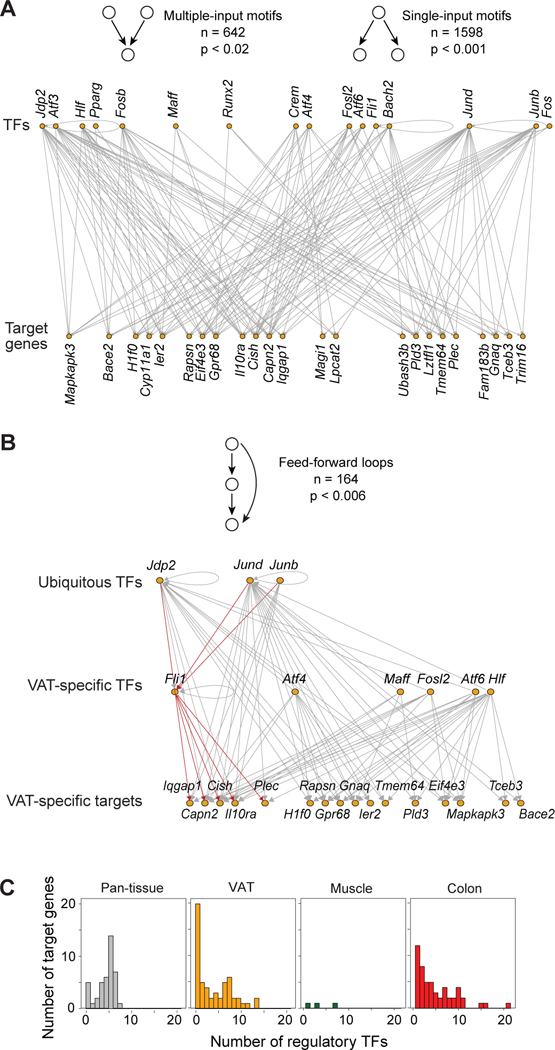
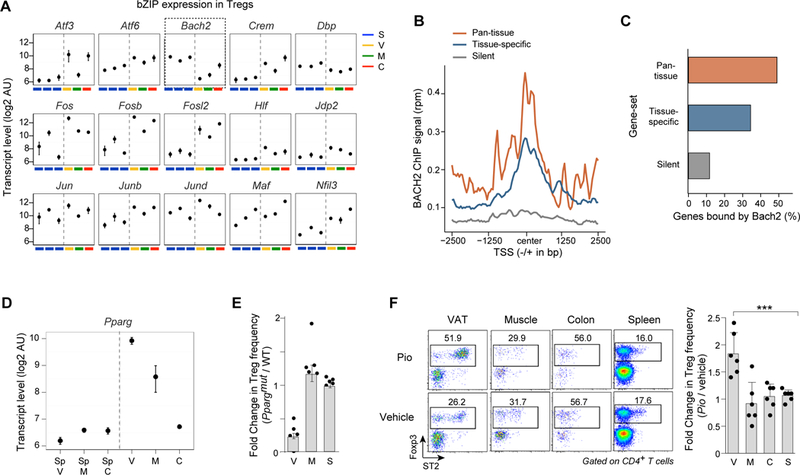
Foxp3+CD4+ regulatory T cells (Tregs) accumulate in certain nonlymphoid tissues, where they control diverse aspects of organ homeostasis. Populations of tissue Tregs, as they have been termed, have transcriptomes distinct from those of their counterparts in lymphoid organs and other nonlymphoid tissues. We examined the diversification of Tregs in visceral adipose tissue, skeletal muscle, and the colon vis-à-vis lymphoid organs from the same individuals. The unique transcriptomes of the various tissue Treg populations resulted from layering of tissue-restricted open chromatin regions over regions already open in the spleen, the latter tagged by super-enhancers and particular histone marks. The binding motifs for a small number of transcription factor (TF) families were repeatedly enriched within the accessible chromatin stretches of Tregs in the three nonlymphoid tissues. However, a bioinformatically and experimentally validated transcriptional network, constructed by integrating chromatin accessibility and single-cell transcriptomic data, predicted reliance on different TF family members in the different tissues. The network analysis also revealed that tissue-restricted and broadly acting TFs were integrated into feed-forward loops to enforce tissue-specific gene expression in nonlymphoid-tissue Tregs. Overall, this study provides a framework for understanding the epigenetic dynamics of T cells operating in nonlymphoid tissues, which should inform strategies for specifically targeting them.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Competing interests:
the authors declare that they have no competing interests.
Figures
Similar articles
-
PPARγ marks splenic precursors of multiple nonlymphoid-tissue Treg compartments.Proc Natl Acad Sci U S A. 2021 Mar 30;118(13):e2025197118. doi: 10.1073/pnas.2025197118. Proc Natl Acad Sci U S A. 2021. PMID: 33753509 Free PMC article.
-
TCR Transgenic Mice Reveal Stepwise, Multi-site Acquisition of the Distinctive Fat-Treg Phenotype.Cell. 2018 Jul 12;174(2):285-299.e12. doi: 10.1016/j.cell.2018.05.004. Epub 2018 Jun 7. Cell. 2018. PMID: 29887374 Free PMC article.
-
Tissue Tregs.Annu Rev Immunol. 2016 May 20;34:609-33. doi: 10.1146/annurev-immunol-032712-095948. Annu Rev Immunol. 2016. PMID: 27168246 Free PMC article. Review.
-
Tissue regulatory T cells: regulatory chameleons.Nat Rev Immunol. 2021 Sep;21(9):597-611. doi: 10.1038/s41577-021-00519-w. Epub 2021 Mar 26. Nat Rev Immunol. 2021. PMID: 33772242 Free PMC article. Review.
-
Derivation and Differentiation of Adipose-Tissue Regulatory T Cells: A Stepwise, Multi-Site Process.Front Immunol. 2020 Oct 29;11:599277. doi: 10.3389/fimmu.2020.599277. eCollection 2020. Front Immunol. 2020. PMID: 33193452 Free PMC article. Review.
Cited by
-
An integrated transcription factor framework for Treg identity and diversity.Proc Natl Acad Sci U S A. 2024 Sep 3;121(36):e2411301121. doi: 10.1073/pnas.2411301121. Epub 2024 Aug 28. Proc Natl Acad Sci U S A. 2024. PMID: 39196621 Free PMC article.
-
Tregs in visceral adipose tissue up-regulate circadian-clock expression to promote fitness and enforce a diurnal rhythm of lipolysis.Sci Immunol. 2022 Sep 30;7(75):eabl7641. doi: 10.1126/sciimmunol.abl7641. Epub 2022 Sep 30. Sci Immunol. 2022. PMID: 36179011 Free PMC article.
-
Tissue Tregs and Maintenance of Tissue Homeostasis.Front Cell Dev Biol. 2021 Aug 18;9:717903. doi: 10.3389/fcell.2021.717903. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34490267 Free PMC article. Review.
-
Deciphering the developmental trajectory of tissue-resident Foxp3+ regulatory T cells.Front Immunol. 2024 Mar 28;15:1331846. doi: 10.3389/fimmu.2024.1331846. eCollection 2024. Front Immunol. 2024. PMID: 38605970 Free PMC article. Review.
-
Insights into phenotypic and functional CD8+ TRM heterogeneity.Immunol Rev. 2023 Jul;316(1):8-22. doi: 10.1111/imr.13218. Epub 2023 May 16. Immunol Rev. 2023. PMID: 37191051 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous