

PLK1 Inhibition Targets Myc-Activated Malignant Glioma Cells Irrespective of Mismatch Repair Deficiency-Mediated Acquired Resistance to Temozolomide
- PMID: 30217967
- PMCID: PMC6279590
- DOI: 10.1158/1535-7163.MCT-18-0177
PLK1 Inhibition Targets Myc-Activated Malignant Glioma Cells Irrespective of Mismatch Repair Deficiency-Mediated Acquired Resistance to Temozolomide
Abstract
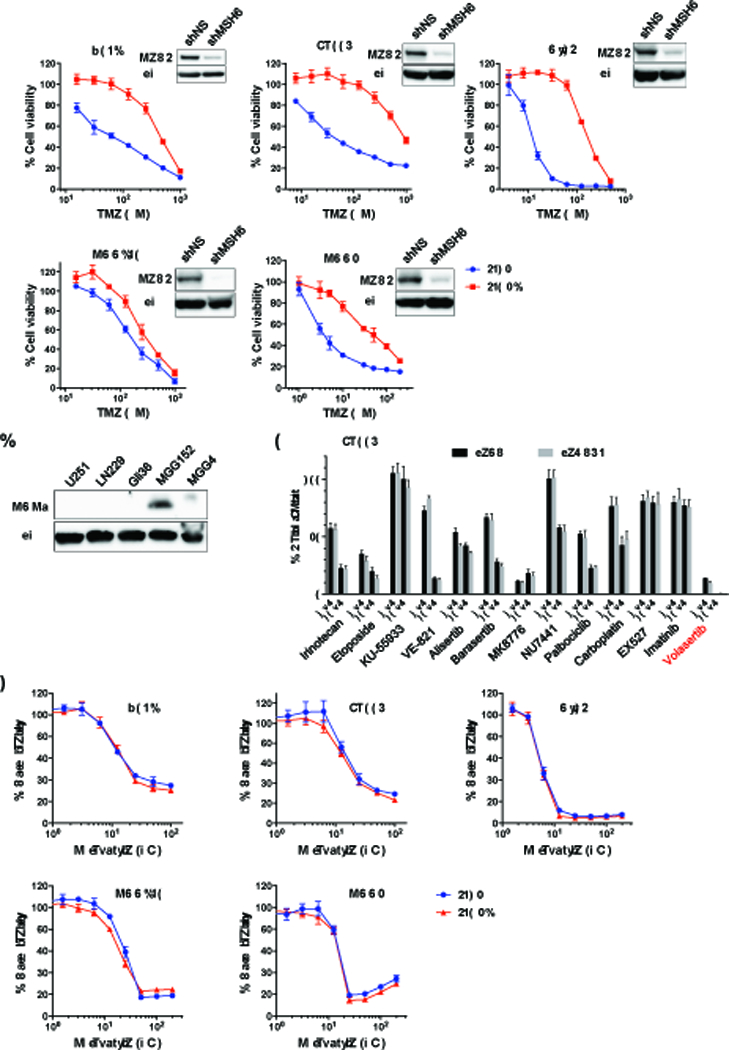
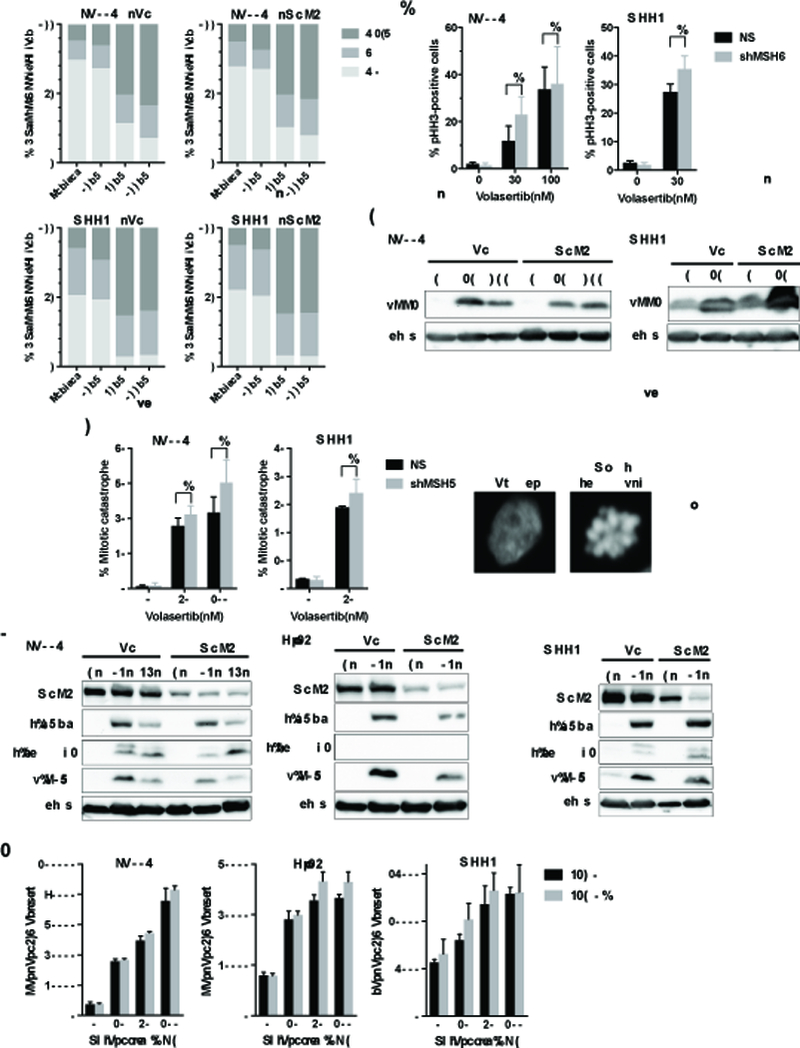
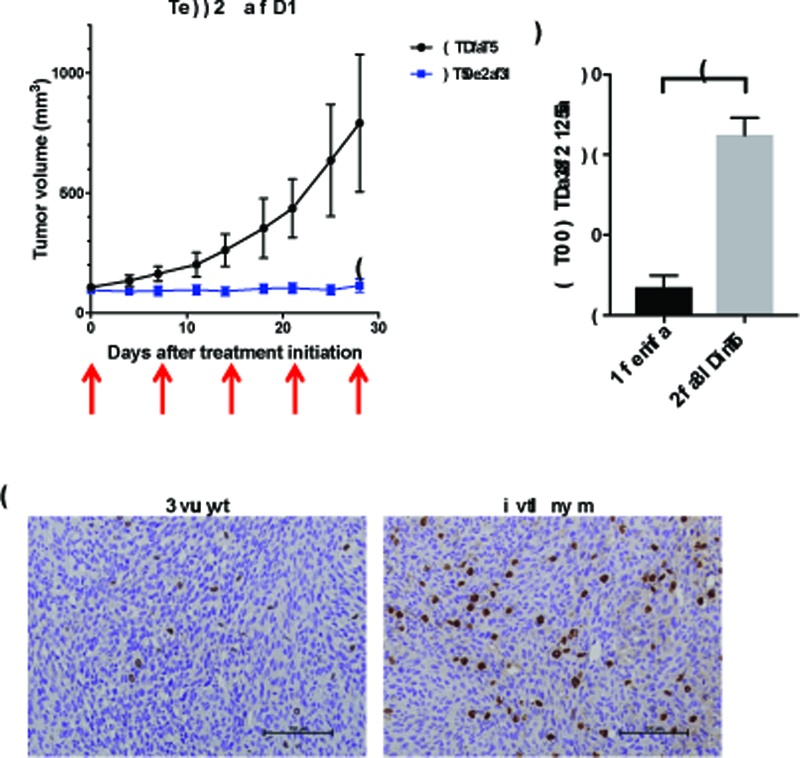
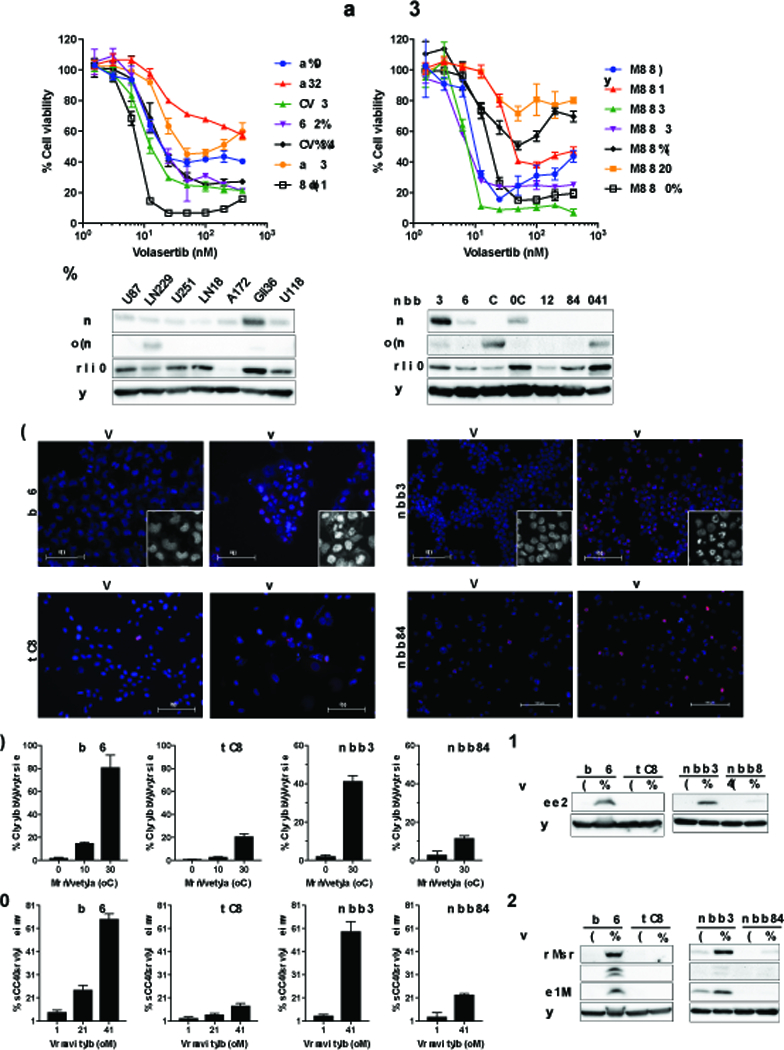
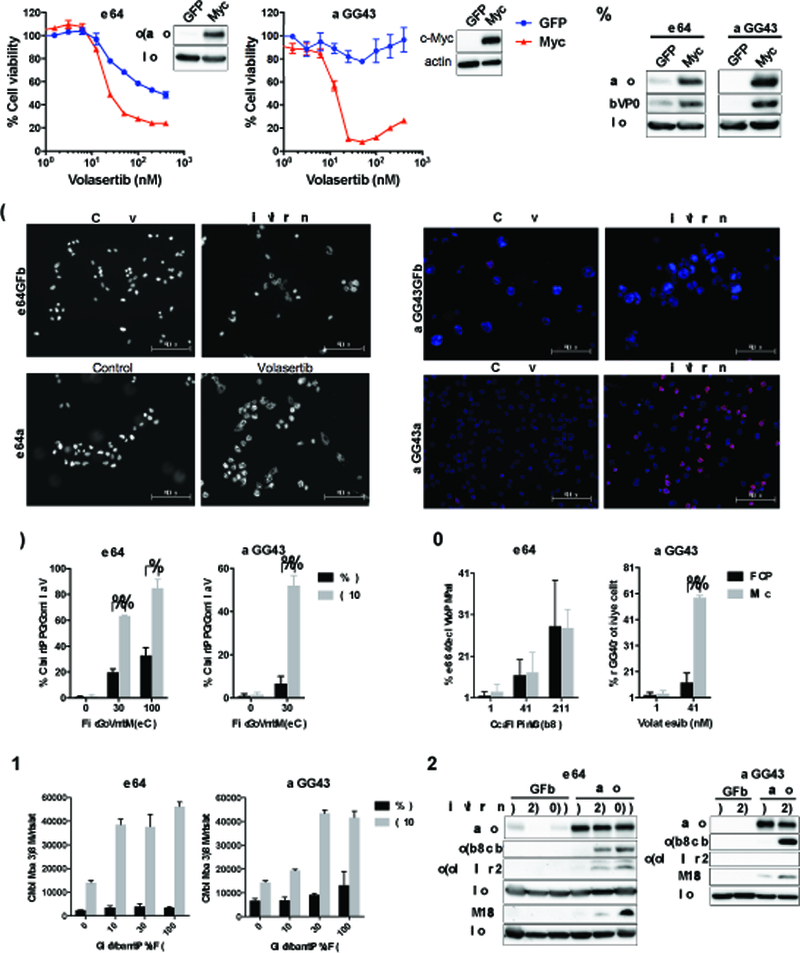
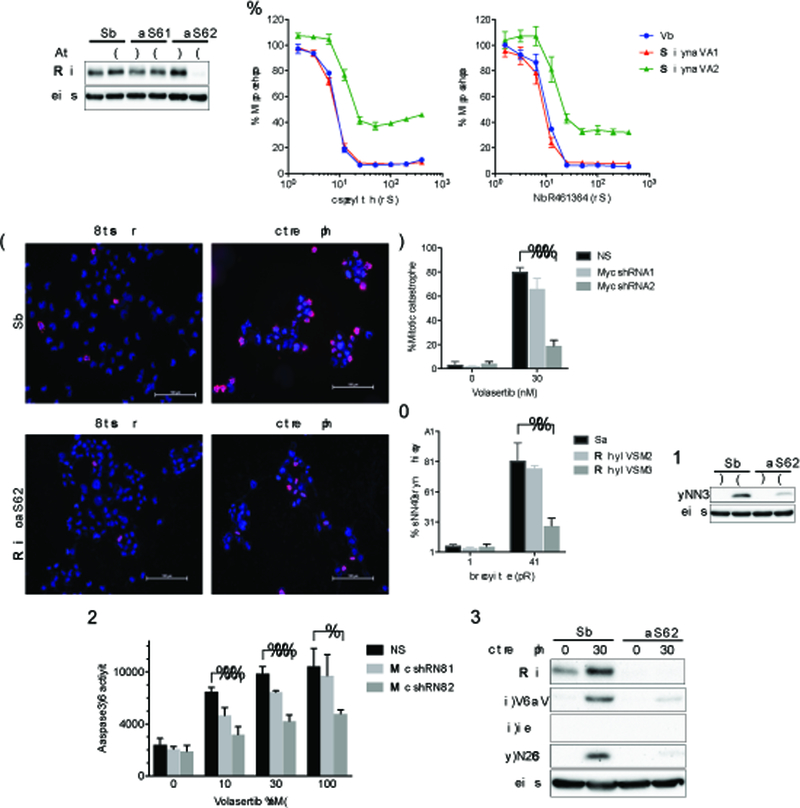
Mismatch repair (MMR) deficiency through MSH6 inactivation has been identified in up to 30% of recurrent high-grade gliomas, and represents a key molecular mechanism underlying the acquired resistance to the alkylating agent temozolomide (TMZ). To develop a therapeutic strategy that could be effective in these TMZ-refractory gliomas, we first screened 13 DNA damage response modulators for their ability to suppress viability of MSH6-inactivated, TMZ-resistant glioma cells. We identified a PLK1 selective inhibitor, Volasertib, as the most potent in inhibiting proliferation of glioblastoma cells. PLK1 inhibition induced mitotic catastrophe, G2-M cell-cycle arrest, and DNA damage, leading to caspase-mediated apoptosis in glioblastoma cells. Importantly, therapeutic effects of PLK1 inhibitors were not influenced by MSH6 knockdown, indicating that their action is independent of MMR status of the cells. Systemic treatment with Volasertib potently inhibited tumor growth in an MMR-deficient, TMZ-resistant glioblastoma xenograft model. Further in vitro testing in established and patient-derived cell line panels revealed an association of PLK1 inhibitor efficacy with cellular Myc expression status. We found that cells with deregulated Myc are vulnerable to PLK1 inhibition, as Myc overexpression sensitizes, whereas its silencing desensitizes, glioblastoma cells to PLK1 inhibitors. This discovery is clinically relevant as glioma progression post-TMZ treatment is frequently accompanied by MYC genomic amplification and/or pathway activation. In conclusion, PLK inhibitor represents a novel therapeutic option for recurrent gliomas, including those TMZ-resistant from MMR deficiency. Genomic MYC alteration may serve as a biomarker for PLK inhibitor sensitivity, as Myc-driven tumors demonstrated pronounced responses.
©2018 American Association for Cancer Research.
Conflict of interest statement
Conflicts of Interest:
All authors have no conflicts of interest to report with regard to this manuscript.
Figures
References
-
- van den Bent MJ, Baumert B, Erridge SC, Vogelbaum MA, Nowak AK, Sanson M, et al. Interim results from the CATNON trial (EORTC study 26053–22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: a phase 3, randomised, open-label intergroup study. Lancet. 2017;390:1645–53. - PMC - PubMed
-
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–96. - PubMed
-
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology. 2009;10:459–66. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
