

ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants
- PMID: 30220433
- PMCID: PMC6174354
- DOI: 10.1016/j.ajhg.2018.08.005
ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants
Abstract
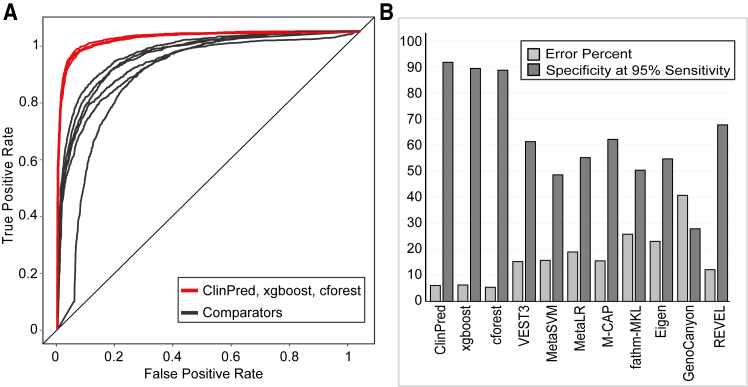
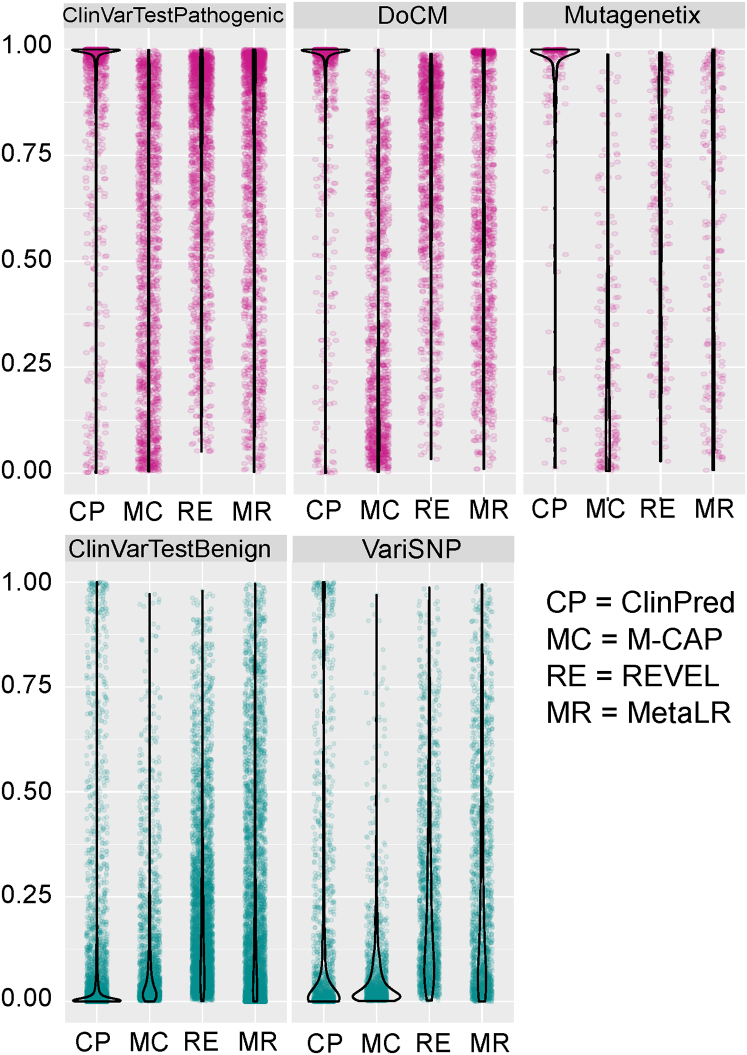
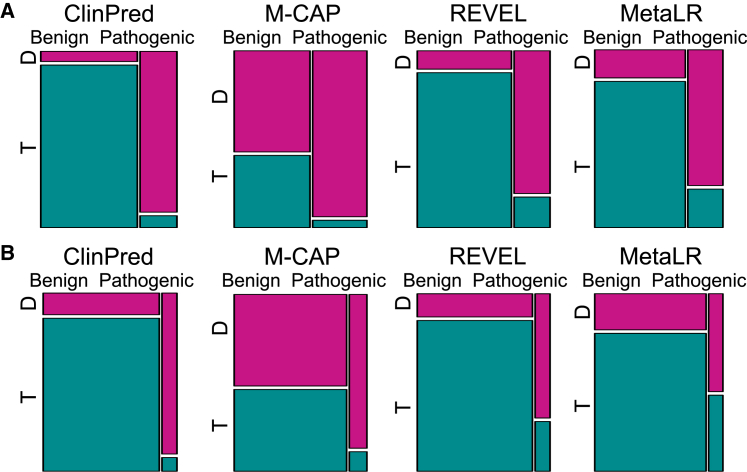
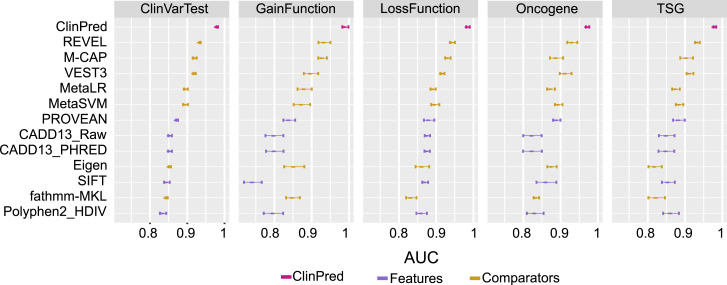
Advances in high-throughput DNA sequencing have revolutionized the discovery of variants in the human genome; however, interpreting the phenotypic effects of those variants is still a challenge. While several computational approaches to predict variant impact are available, their accuracy is limited and further improvement is needed. Here, we introduce ClinPred, an efficient tool for identifying disease-relevant nonsynonymous variants. Our predictor incorporates two machine learning algorithms that use existing pathogenicity scores and, notably, benefits from inclusion of normal population allele frequency from the gnomAD database as an input feature. Another major strength of our approach is the use of ClinVar-a rapidly growing database that allows selection of confidently annotated disease-causing variants-as a training set. Compared to other methods, ClinPred showed superior accuracy for predicting pathogenicity, achieving the highest area under the curve (AUC) score and increasing both the specificity and sensitivity in different test datasets. It also obtained the best performance according to various other metrics. Moreover, ClinPred performance remained robust with respect to disease type (cancer or rare disease) and mechanism (gain or loss of function). Importantly, we observed that adding allele frequency as a predictive feature-as opposed to setting fixed allele frequency cutoffs-boosts the performance of prediction. We provide pre-computed ClinPred scores for all possible human missense variants in the exome to facilitate its use by the community.
Keywords: cancer; computational biology; diagnostic; machine learning; pathogenicity prediction; predictive modeling; rare disease; variant interpretation; whole-exome sequencing.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
