

Inhibition of Atypical Protein Kinase C Reduces Inflammation-Induced Retinal Vascular Permeability
- PMID: 30220554
- PMCID: PMC6180272
- DOI: 10.1016/j.ajpath.2018.06.020
Inhibition of Atypical Protein Kinase C Reduces Inflammation-Induced Retinal Vascular Permeability
Abstract
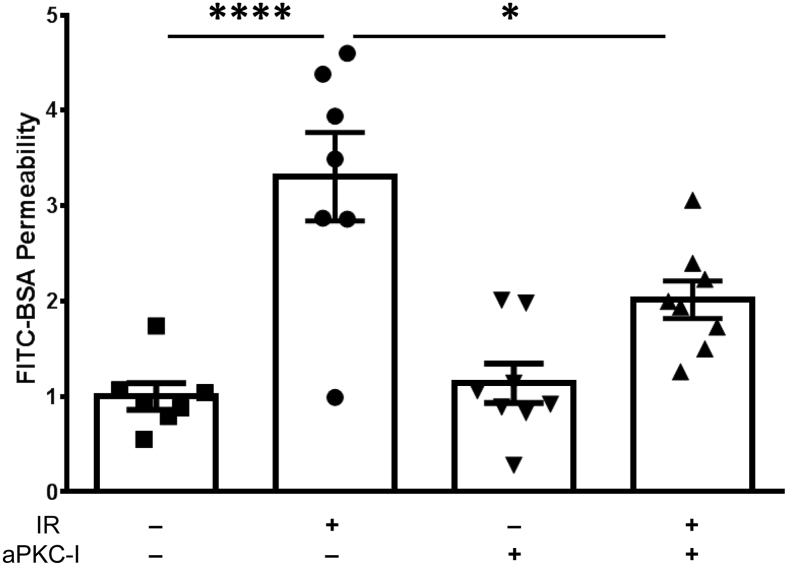
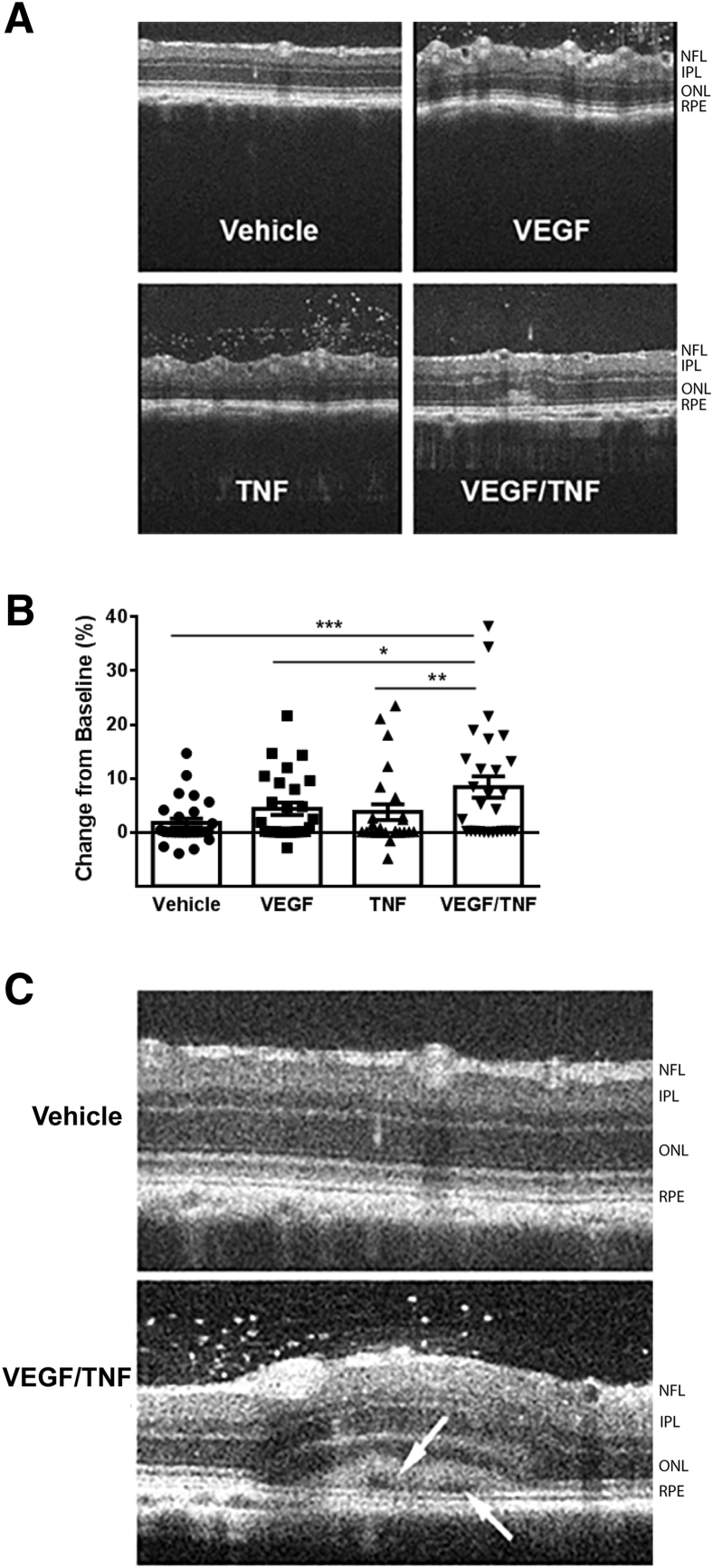
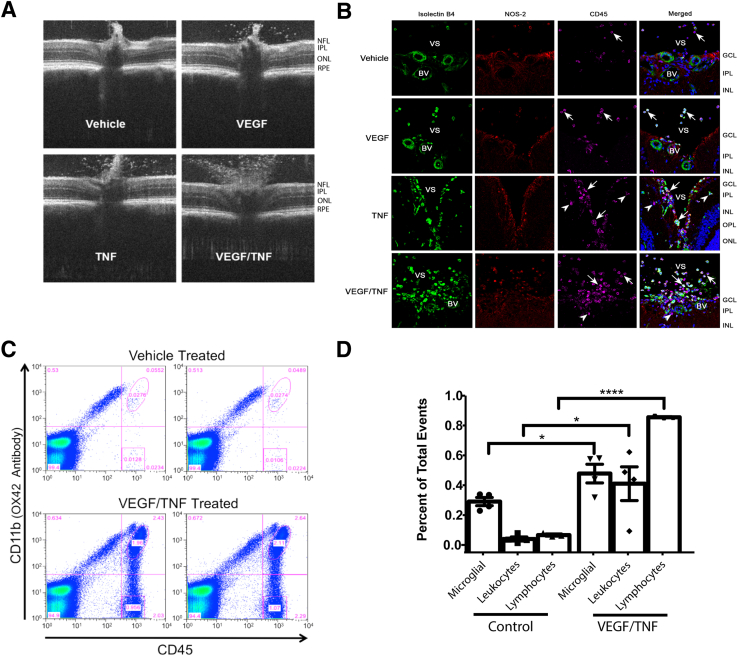
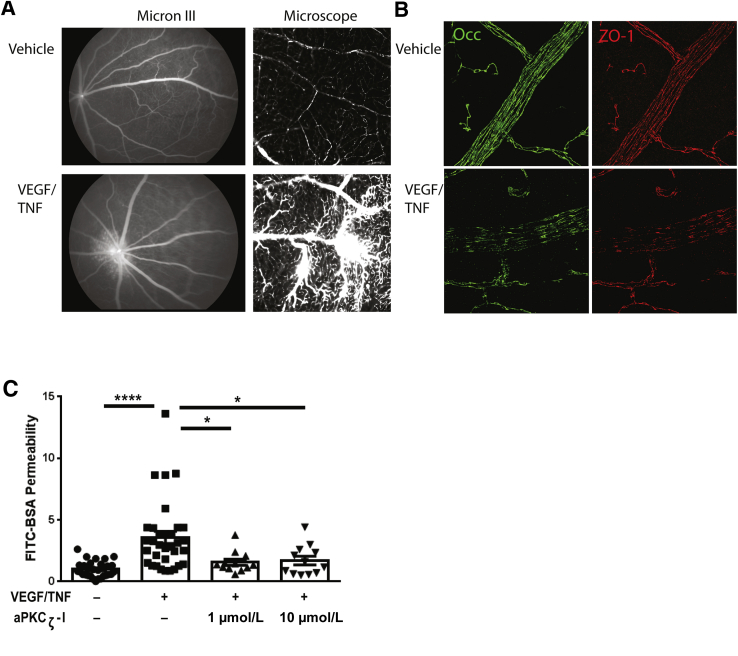
Changes in permeability of retinal blood vessels contribute to macular edema and the pathophysiology of numerous ocular diseases, including diabetic retinopathy, retinal vein occlusions, and macular degeneration. Vascular endothelial growth factor (VEGF) induces retinal permeability and macular thickening in these diseases. However, inflammatory agents, such as tumor necrosis factor-α (TNF-α), also may drive vascular permeability, specifically in patients unresponsive to anti-VEGF therapy. Recent evidence suggests VEGF and TNF-α induce permeability through distinct mechanisms; however, both require the activation of atypical protein kinase C (aPKC). We provide evidence, using genetic mouse models and therapeutic intervention with small molecules, that inhibition of aPKC prevented or reduced vascular permeability in animal models of retinal inflammation. Expression of a kinase-dead aPKC transgene, driven by a vascular and hematopoietic restricted promoter, reduced retinal vascular permeability in an ischemia-reperfusion model of retinal injury. This effect was recapitulated with a small-molecule inhibitor of aPKC. Expression of the kinase-dead aPKC transgene dramatically reduced the expression of inflammatory factors and blocked the attraction of inflammatory monocytes and granulocytes after ischemic injury. Coinjection of VEGF with TNF-α was sufficient to induce permeability, edema, and retinal inflammation, and treatment with an aPKC inhibitor prevented VEGF/TNF-α-induced permeability. These data suggest that aPKC contributes to inflammation-driven retinal vascular pathology and may be an attractive target for therapeutic intervention.
Copyright © 2018 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
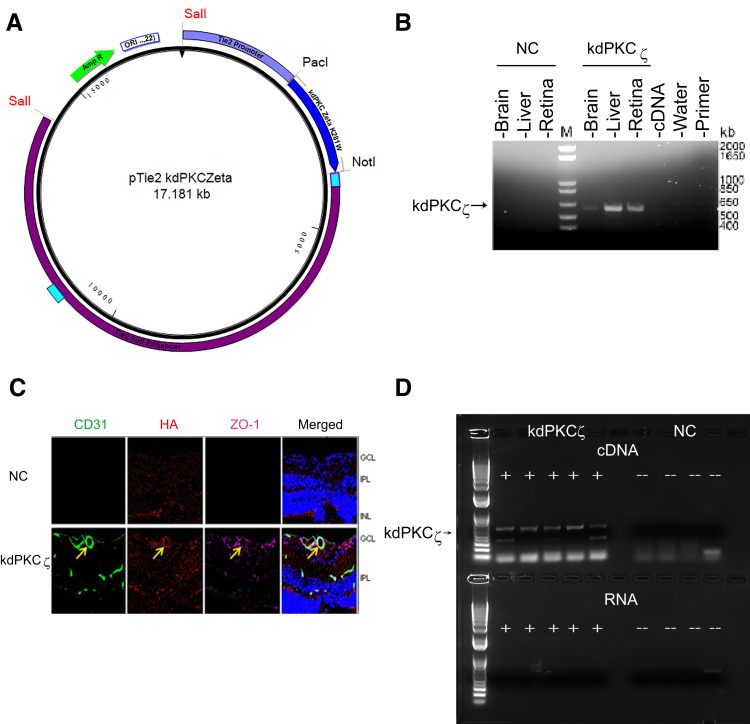
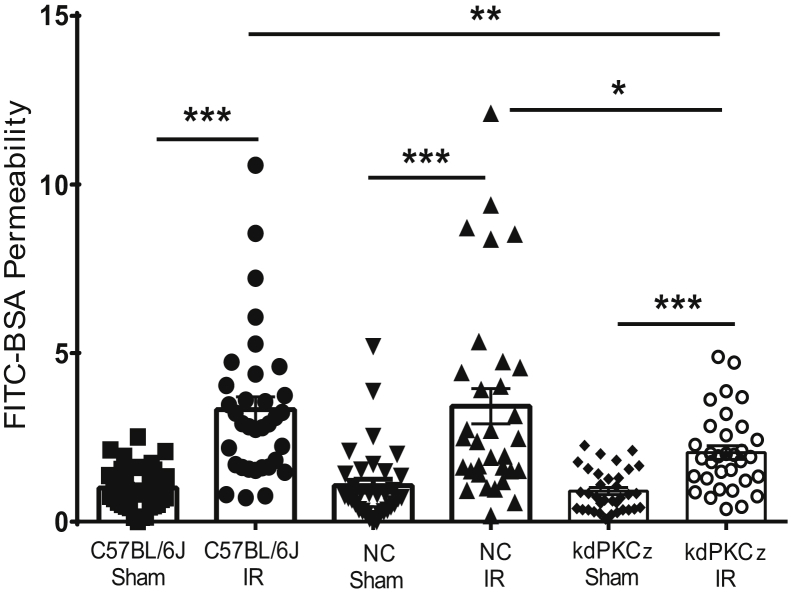
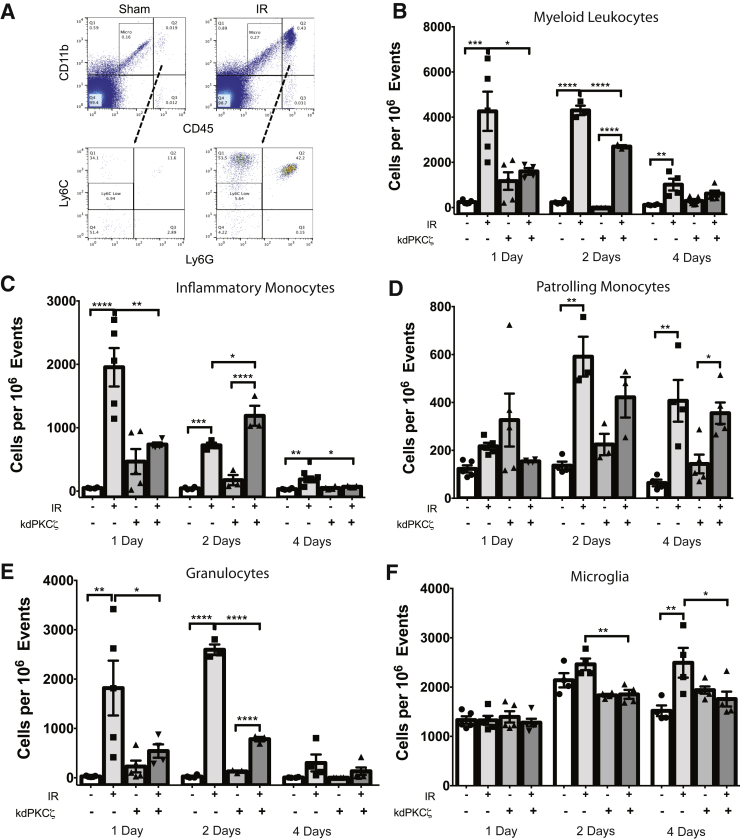
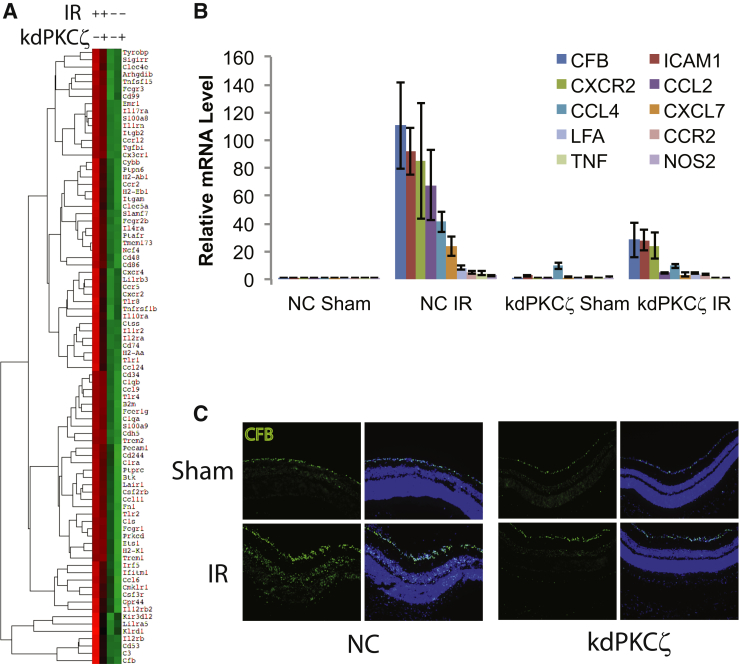
Comment in
-
Atypical Protein Kinase C: Breaking Down Barriers in Ocular Disease?Am J Pathol. 2018 Oct;188(10):2142-2146. doi: 10.1016/j.ajpath.2018.07.006. Epub 2018 Sep 13. Am J Pathol. 2018. PMID: 30220553 Free PMC article.
References
-
- Campochiaro P.A. Anti-vascular endothelial growth factor treatment for retinal vein occlusions. Ophthalmologica. 2012;227 Suppl 1:30–35. - PubMed
-
- Emerson M.V., Lauer A.K. Emerging therapies for the treatment of neovascular age-related macular degeneration and diabetic macular edema. BioDrugs. 2007;21:245–257. - PubMed
-
- Markomichelakis N.N., Theodossiadis P.G., Pantelia E., Papaefthimiou S., Theodossiadis G.P., Sfikakis P.P. Infliximab for chronic cystoid macular edema associated with uveitis. Am J Ophthalmol. 2004;138:648–650. - PubMed
-
- Antonetti D.A., Klein R., Gardner T.W. Diabetic retinopathy. N Engl J Med. 2012;366:1227–1239. - PubMed
-
- Johnson M.W. Etiology and treatment of macular edema. Am J Ophthalmol. 2009;147:11–21.e1. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
