

Proteome-Wide Structural Biology: An Emerging Field for the Structural Analysis of Proteins on the Proteomic Scale
- PMID: 30222357
- PMCID: PMC6524533
- DOI: 10.1021/acs.jproteome.8b00341
Proteome-Wide Structural Biology: An Emerging Field for the Structural Analysis of Proteins on the Proteomic Scale
Abstract
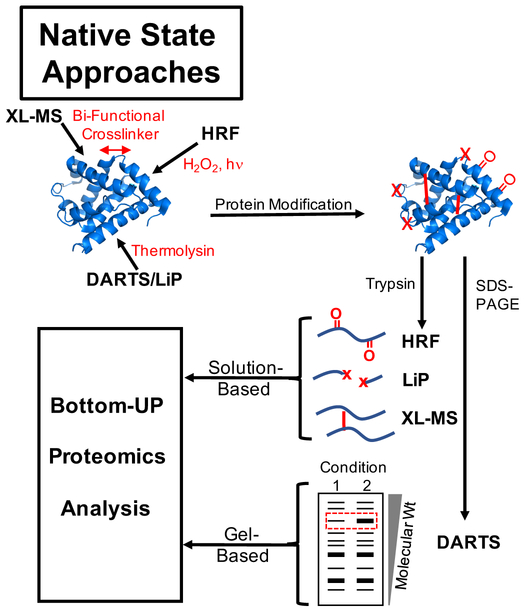
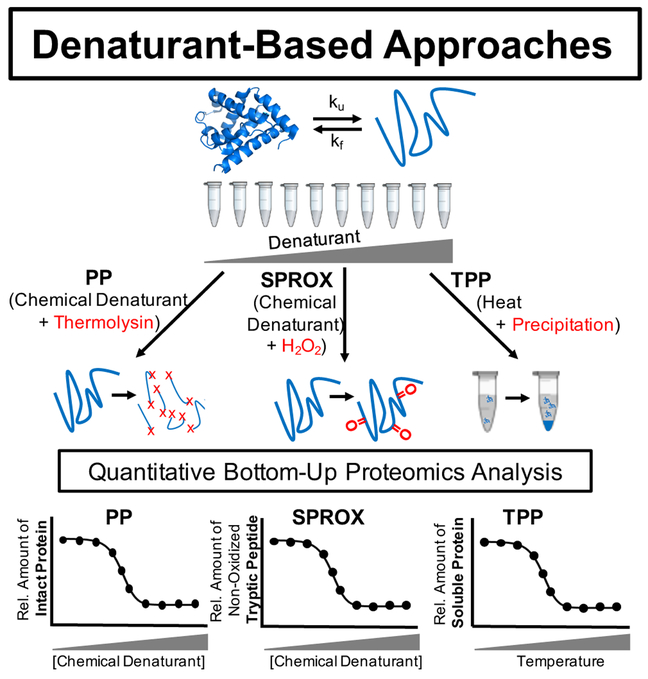
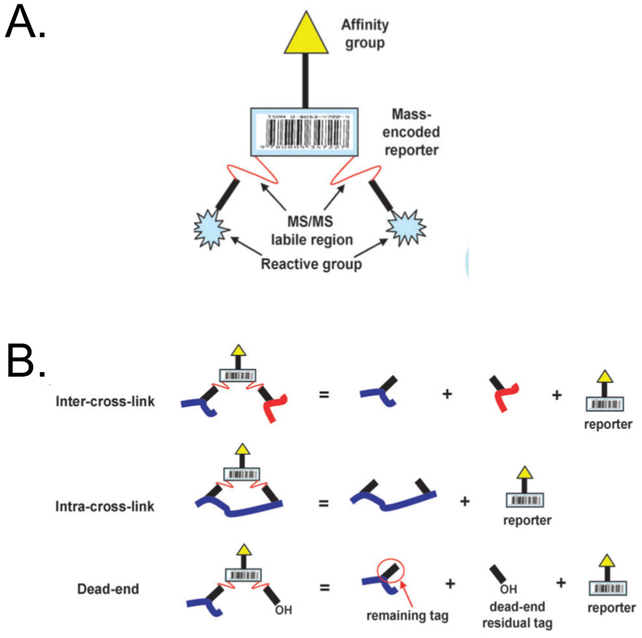
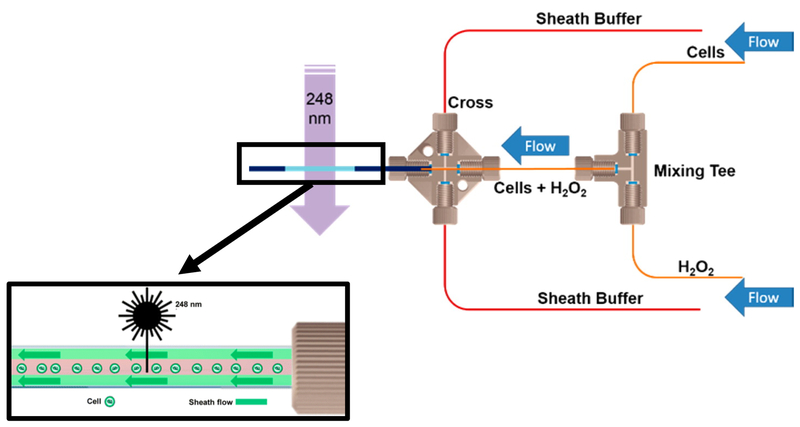
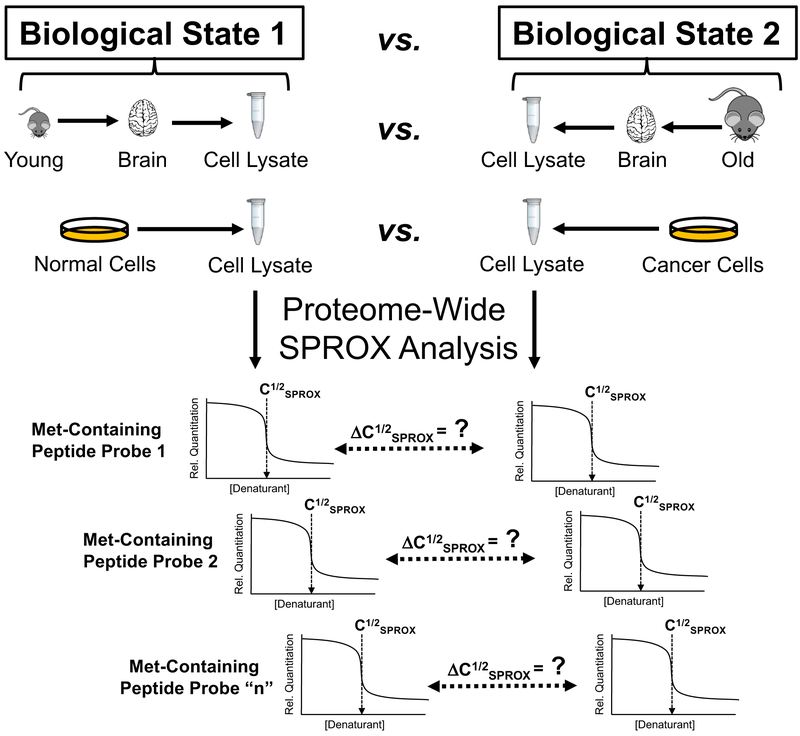
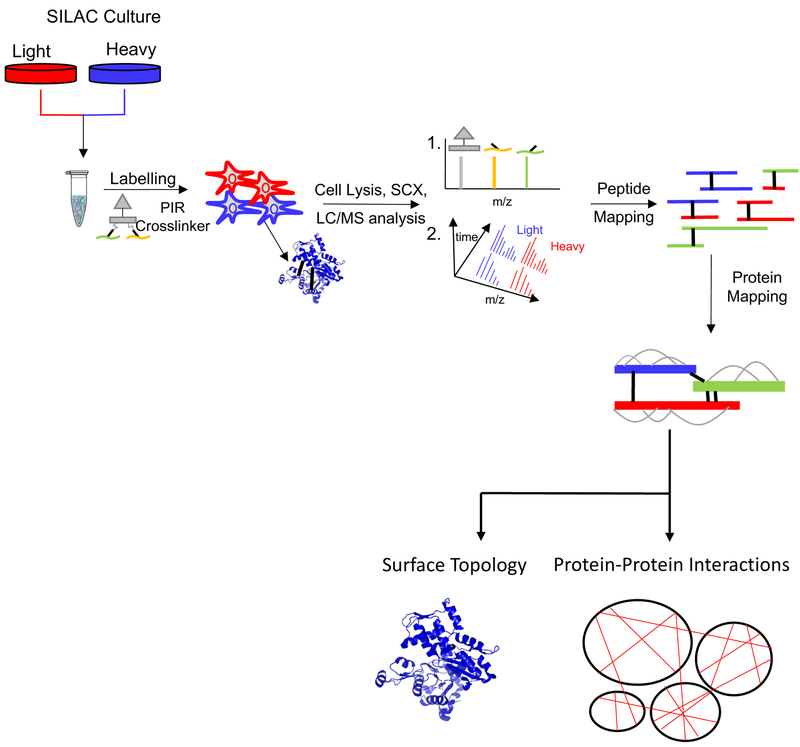
Over the past decade, a suite of new mass-spectrometry-based proteomics methods has been developed that now enables the conformational properties of proteins and protein-ligand complexes to be studied in complex biological mixtures, from cell lysates to intact cells. Highlighted here are seven of the techniques in this new toolbox. These techniques include chemical cross-linking (XL-MS), hydroxyl radical footprinting (HRF), Drug Affinity Responsive Target Stability (DARTS), Limited Proteolysis (LiP), Pulse Proteolysis (PP), Stability of Proteins from Rates of Oxidation (SPROX), and Thermal Proteome Profiling (TPP). The above techniques all rely on conventional bottom-up proteomics strategies for peptide sequencing and protein identification. However, they have required the development of unconventional proteomic data analysis strategies. Discussed here are the current technical challenges associated with these different data analysis strategies as well as the relative analytical capabilities of the different techniques. The new biophysical capabilities that the above techniques bring to bear on proteomic research are also highlighted in the context of several different application areas in which these techniques have been used, including the study of protein ligand binding interactions (e.g., protein target discovery studies and protein interaction network analyses) and the characterization of biological states.
Keywords: mass spectrometry; protein folding; proteomics; thermodynamics.
Figures
Similar articles
-
Innovation: Structural Proteomics Goes Global.J Proteome Res. 2018 Nov 2;17(11):3613. doi: 10.1021/acs.jproteome.8b00698. Epub 2018 Oct 10. J Proteome Res. 2018. PMID: 30303008 No abstract available.
-
SILAC-pulse proteolysis: A mass spectrometry-based method for discovery and cross-validation in proteome-wide studies of ligand binding.J Am Soc Mass Spectrom. 2014 Dec;25(12):2073-83. doi: 10.1007/s13361-014-0992-y. Epub 2014 Oct 15. J Am Soc Mass Spectrom. 2014. PMID: 25315461
-
Mass spectrometry analysis of the structural proteome.Curr Opin Struct Biol. 2020 Feb;60:57-65. doi: 10.1016/j.sbi.2019.10.006. Epub 2019 Dec 13. Curr Opin Struct Biol. 2020. PMID: 31841731 Review.
-
Painting proteins with covalent labels: what's in the picture?J Am Soc Mass Spectrom. 2009 Jun;20(6):1193-206. doi: 10.1016/j.jasms.2009.02.006. Epub 2009 Feb 12. J Am Soc Mass Spectrom. 2009. PMID: 19269190
-
Mass spectrometry-based methods for structural biology on a proteome-wide scale.Biochem Soc Trans. 2020 Jun 30;48(3):945-954. doi: 10.1042/BST20190794. Biochem Soc Trans. 2020. PMID: 32412054 Review.
Cited by
-
Current Advances in CETSA.Front Mol Biosci. 2022 Jun 9;9:866764. doi: 10.3389/fmolb.2022.866764. eCollection 2022. Front Mol Biosci. 2022. PMID: 35755818 Free PMC article. Review.
-
Illuminating Biological Interactions with in Vivo Protein Footprinting.Anal Chem. 2019 May 21;91(10):6577-6584. doi: 10.1021/acs.analchem.9b00244. Epub 2019 May 7. Anal Chem. 2019. PMID: 31025855 Free PMC article.
-
Comparative Analysis of Mass-Spectrometry-Based Proteomic Methods for Protein Target Discovery Using a One-Pot Approach.J Am Soc Mass Spectrom. 2020 Feb 5;31(2):217-226. doi: 10.1021/jasms.9b00041. Epub 2019 Nov 22. J Am Soc Mass Spectrom. 2020. PMID: 32031398 Free PMC article.
-
Cellular Exposure to Chloroacetanilide Herbicides Induces Distinct Protein Destabilization Profiles.ACS Chem Biol. 2023 Jul 21;18(7):1661-1676. doi: 10.1021/acschembio.3c00338. Epub 2023 Jul 10. ACS Chem Biol. 2023. PMID: 37427419 Free PMC article.
-
Standard Protocols for Characterising Primary and In Vitro-Generated Human Hepatocytes.J Cell Mol Med. 2025 Feb;29(3):e70390. doi: 10.1111/jcmm.70390. J Cell Mol Med. 2025. PMID: 39910642 Free PMC article. Review.
References
-
- Christiansen A; Wang Q; Samiotakis A; Cheung MS; Wittung-Stafshede P, Factors defining effects of macromolecular crowding on protein stability: an in vitro/in silico case study using cytochrome c. Biochemistry 2010, 49,(31), 6519–30. - PubMed
-
- Wirth AJ; Gruebele M, Quinary protein structure and the consequences of crowding in living cells: leaving the test-tube behind. Bioessays 2013, 35, (11), 984–93. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
