

Association mapping by aerial drone reveals 213 genetic associations for Sorghum bicolor biomass traits under drought
- PMID: 30223789
- PMCID: PMC6142696
- DOI: 10.1186/s12864-018-5055-5
Association mapping by aerial drone reveals 213 genetic associations for Sorghum bicolor biomass traits under drought
Abstract
Background: Sorghum bicolor is the fifth most commonly grown cereal worldwide and is remarkable for its drought and abiotic stress tolerance. For these reasons and the large size of biomass varieties, it has been proposed as a bioenergy crop. However, little is known about the genes underlying sorghum's abiotic stress tolerance and biomass yield.
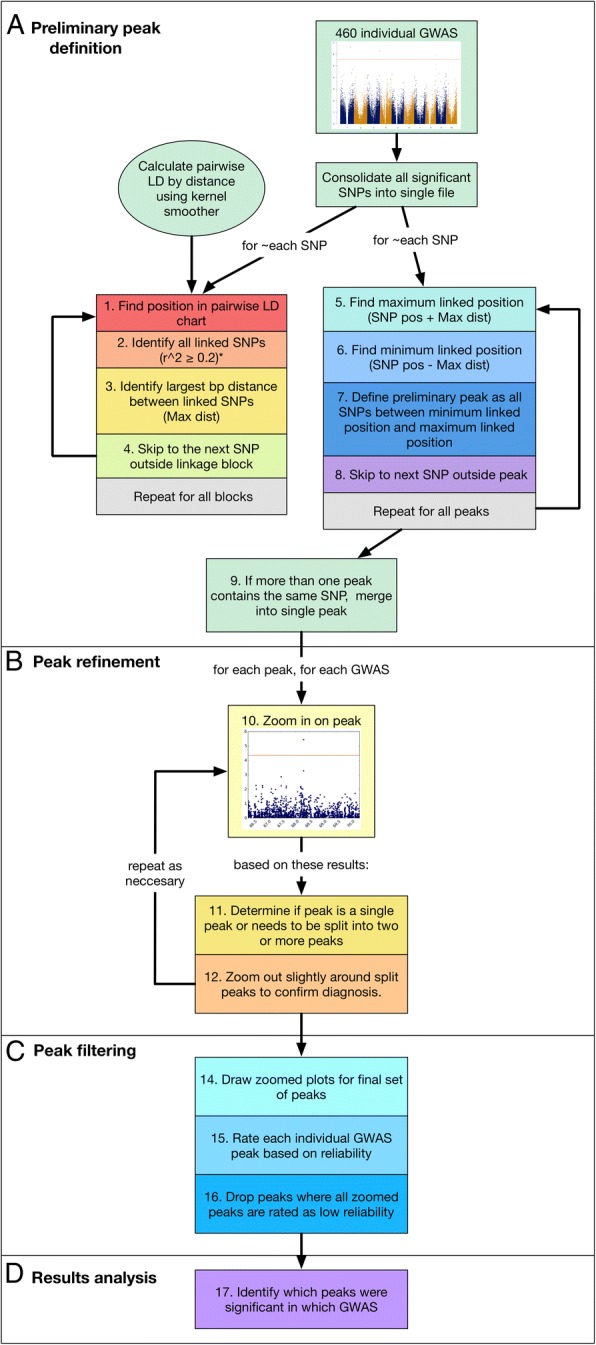
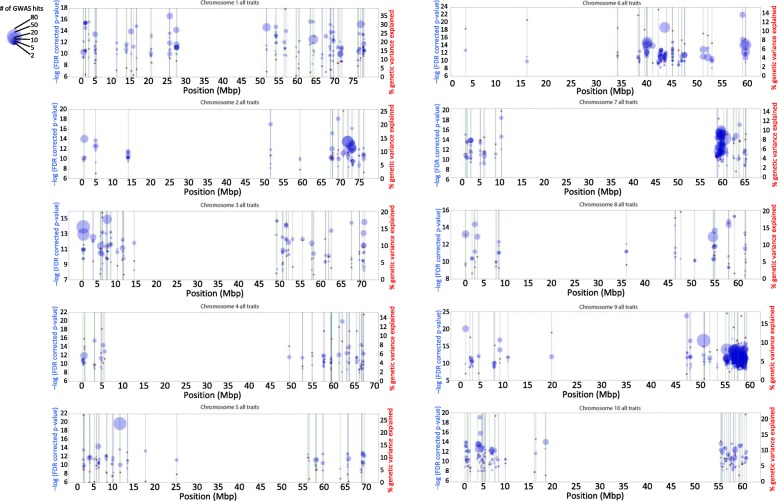
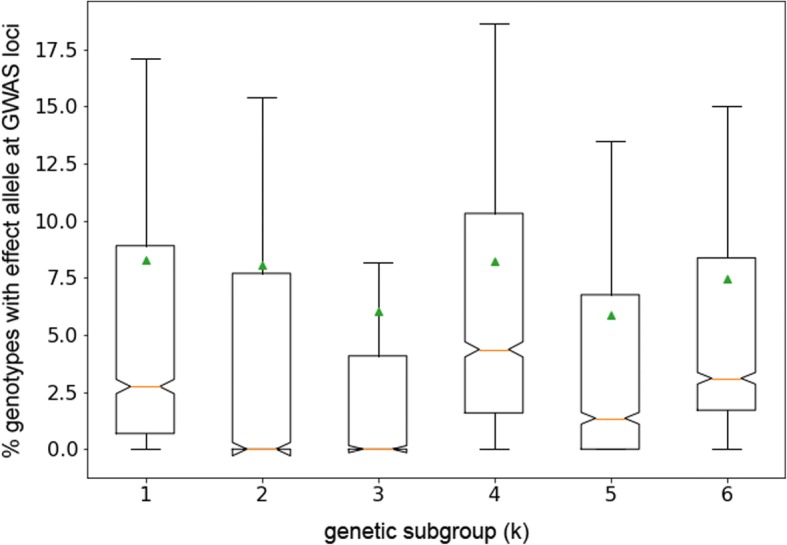
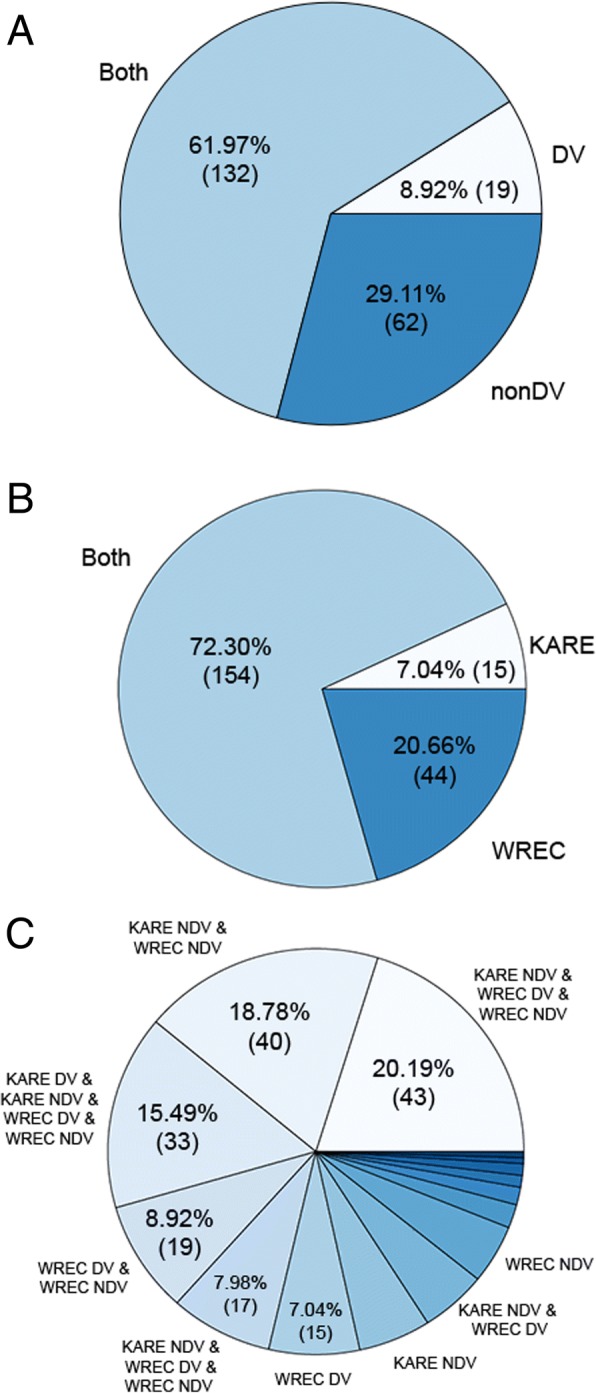
Results: To uncover the genetic basis of drought tolerance in sorghum at a genome-wide level, we undertook a high-density phenomics genome wide association study (GWAS) in which 648 diverse sorghum lines were phenotyped at two locations in California once per week by drone over the course of a growing season. Biomass, height, and leaf area were measured by drone for individual field plots, subjected to two drought treatments and a well-watered control. The resulting dataset of ~ 171,000 phenotypic data-points was analyzed along with 183,989 genotype by sequence markers to reveal 213 high-quality, replicated, and conserved GWAS associations.
Conclusions: The genomic intervals defined by the associations include many strong candidate genes, including those encoding heat shock proteins, antifreeze proteins, and other domains recognized as important to plant stress responses. The markers identified by our study can be used for marker assisted selection for drought tolerance and biomass. In addition, our results are a significant step toward identifying specific sorghum genes controlling drought tolerance and biomass yield.
Keywords: Biomass; Drone; Drought; GWAS; Phenomics; Sorghum.
Conflict of interest statement
Ethics approval and consent to participate
Seeds were obtained from the USDA-ARS Plant Genetic Resources Conservation Unit and increased at UC-ANR-KARE in Parlier, CA and by Chromatin, Inc. at winter facilities in Puerto Rico. No field permission was required to collect plant samples.
Consent for publication
Not applicable.
Competing interests
M.C. works for Blue River Technology which plans to provide the drone phenotyping technology utilized in this study as a commercial service. S.S. works for Chromatin Inc., which seeks to commercialize some of the biomass Sorghum lines included in the research project. All other authors declare no competing financial interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
-
- Cline WR. Global warming and agriculture: End-of-century estimates by country. Washington D.C.: Peterson Institute; 2007.
-
- Rooney WL, Blumenthal J, Bean B, Mullet JE. Designing sorghum as a dedicated bioenergy feedstock. Biofuels Bioprod Biorefin. 2007;1:147–157. doi: 10.1002/bbb.15. - DOI
-
- Saballos A. In: Genetic Improvement of Bioenergy Crops. Vermerris W, editor. New York: Springer; 2008. pp. 211–248.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
